The DNA Damage Response
Our lab has had a long history of studying how DNA damage and DNA replication blocks are sensed and how this information is relayed throughout the cell to profoundly alter cellular physiology and to promote DNA repair and cell survival. Cells are basically blind and when undergoing complex repairs such as at DNA replication forks, they must be able to feel all the relevant structures and communicate their presence to the repair machinery in order to resolve any and all such problems. We are continuing to uncover new components of this complex pathway and hope to integrate this knowledge with and understanding of the role of these pathways in cancer therapies.
- Recent Studies
- The Protexin complex counters resection on stalled forks to promote homologous recombination and crosslink repair
- The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4
- Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program
- Older Work
Recent Studies
The Protexin complex counters resection on stalled forks to promote homologous recombination and crosslink repair
Protection of stalled replication forks is critical to genomic stability. Using genetic and proteomic analyses, we discovered the Protexin complex containing the ssDNA binding protein SCAI and the DNA polymerase REV3. Protexin is required specifically for protecting forks stalled by nucleotide depletion, fork barriers, fragile sites, and DNA inter-strand crosslinks (ICLs), where it promotes homologous recombination and repair. Protexin loss leads to ssDNA accumulation and profound genomic instability in response to ICLs. Protexin interacts with RNA POL2, and both oppose EXO1’s resection of DNA on forks remodeled by the FANCM translocase activity. This pathway acts independently of BRCA/RAD51-mediated fork stabilization, and cells with BRCA2 mutations were dependent on SCAI for survival. These data suggest that Protexin and its associated factors establish a new fork protection pathway that counteracts fork resection in part through a REV3 polymerase-dependent resynthesis mechanism of excised DNA, particularly at ICL stalled forks.

The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4
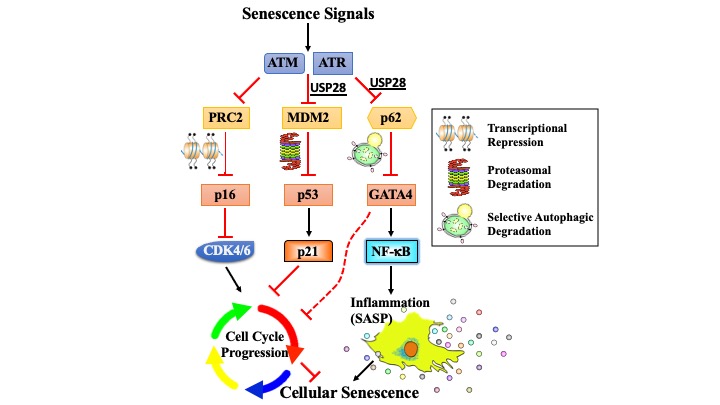
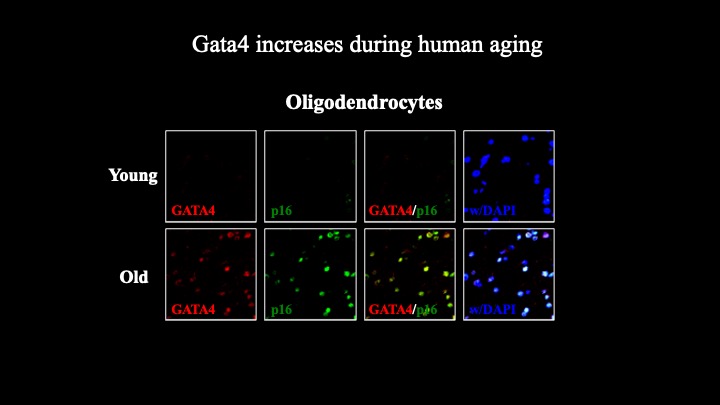
Cellular senescence is a terminal stress-activated program controlled by the p53 and p16(INK4a) tumor suppressor proteins. A striking feature of senescence is the senescence-associated secretory phenotype (SASP), a pro-inflammatory response linked to tumor promotion and aging. We have identified the transcription factor GATA4 as a senescence and SASP regulator. GATA4 is stabilized in cells undergoing senescence and is required for the SASP. Normally, GATA4 is degraded by p62-mediated selective autophagy, but this regulation is suppressed during senescence, thereby stabilizing GATA4. GATA4 in turn activates the transcription factor NF-κB to initiate the SASP and facilitate senescence. GATA4 activation depends on the DNA damage response regulators ATM and ATR, but not on p53 or p16(INK4a). GATA4 accumulates in multiple tissues, including the aging brain, and could contribute to aging and its associated inflammation.
Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program
Senescence is a terminal differentiation program that halts the growth of damaged cells and must be circumvented for cancer to arise. Here we describe a panel of genetic screens to identify genes required for replicative senescence. We uncover a role in senescence for the potent tumor suppressor and ATM substrate USP28. USP28 controls activation of both the TP53 branch and the GATA4/NFkB branch that controls the senescence-associated secretory phenotype (SASP). These results suggest a role for ubiquitination in senescence and imply a common node downstream from ATM that links the TP53 and GATA4 branches of the senescence response.
RFWD3-dependent ubiquitination of RPA regulates repair at stalled replication forks
We have used quantitative proteomics to profile ubiquitination in the DNA damage response (DDR). We demonstrate that RPA, which functions as a protein scaffold in the replication stress response, is multiply ubiquitinated upon replication fork stalling. Ubiquitination of RPA occurs on chromatin, involves sites outside its DNA binding channel, does not cause proteasomal degradation, and increases under conditions of fork collapse, suggesting a role in repair at stalled forks. We demonstrate that the E3 ligase RFWD3 mediates RPA ubiquitination. RFWD3 is necessary for replication fork restart, normal repair kinetics during replication stress, and homologous recombination (HR) at stalled replication forks. Mutational analysis suggests that multisite ubiquitination of the entire RPA complex is responsible for repair at stalled forks. Multisite protein group sumoylation is known to promote HR in yeast. Our findings reveal a similar requirement for multisite protein group ubiquitination during HR at stalled forks in mammalian cells.
Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response
The signal transduction pathway of the DNA damage response (DDR) is activated to maintain genomic integrity following DNA damage. The DDR promotes genomic integrity by regulating a large network of cellular activities that range from DNA replication and repair to transcription, RNA splicing, and metabolism. In this study we define an interaction between the DDR factor NBS1 and TCOF1, a nucleolar protein that regulates ribosomal DNA (rDNA) transcription and is mutated in Treacher Collins syndrome. We show that NBS1 relocalizes to nucleoli after DNA damage in a manner dependent on TCOF1 and on casein kinase II and ATM, which are known to modify TCOF1 by phosphorylation. Moreover, we identify a putative ATM phosphorylation site that is required for NBS1 relocalization to nucleoli in response to DNA damage. Last, we report that TCOF1 promotes cellular resistance to DNA damaging agents. Collectively, our findings identify TCOF1 as a DDR factor that could cooperate with ATM and NBS1 to suppress inappropriate rDNA transcription and maintain genomic integrity after DNA damage.
DNA repair: mechanism of DNA inter-strand crosslink processing by repair nuclease FAN1
DNA interstrand cross-links (ICLs) are highly toxic lesions associated with cancer and degenerative diseases. ICLs can be repaired by the Fanconi anemia (FA) pathway and through FA-independent processes involving the FAN1 nuclease. In this work, FAN1-DNA crystal structures and biochemical data reveal that human FAN1 cleaves DNA successively at every third nucleotide. In vitro, this exonuclease mechanism allows FAN1 to excise an ICL from one strand through flanking incisions. DNA access requires a 5′-terminal phosphate anchor at a nick or a 1- or 2-nucleotide flap and is augmented by a 3′ flap, suggesting that FAN1 action is coupled to DNA synthesis or recombination. FAN1’s mechanism of ICL excision is well suited for processing other localized DNA adducts as well.
Older Work
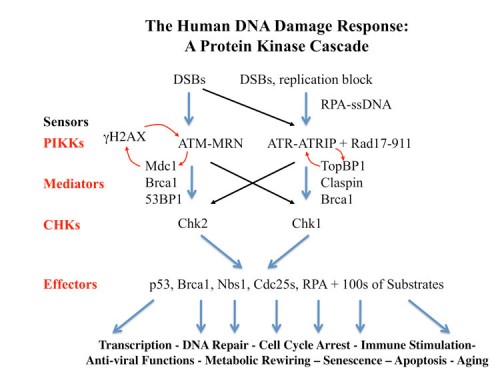
The Human DDR as a Protein Kinase Cascade
Our work on yeast led us into elucidation of the mammalian DNA Damage and DNA replication Stress response pathways. These pathways outlines here, are similar to those we found in yeast but contain more players and carry out metazoan-specific functions not seen in the fungal systems. Below we discuss some of the recent findings.
How DNA Damage and Replication Stress Are Sensed and Activate the DDR
Several years ago we identified the ATR-interacting protein, ATRIP (142), which is an essential component of the DNA damage checkpoint pathway and is required to localize ATR to sites of DNA damage. ATRIP forms a tight complex with ATR and deletion of either protein destabilizes the other. Thus, ATRIP and ATR are mutually dependent partners that work together to transduce the DNA damage signal in mammals.
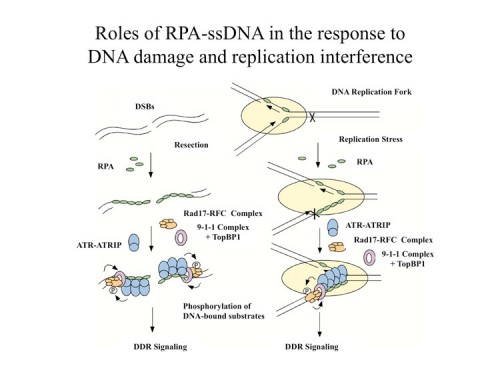
While we knew that ATR/ATRIP localized to sites of DNA damage, we had no idea how DNA damage is sensed. However, we were able to address this question when we discovered that replication protein A (RPA), a protein complex that associates with single-stranded DNA (ssDNA), is required for the recruitment of ATR to sites of DNA damage (166) and for ATR-mediated Chk1 activation in human cells. In vitro, RPA stimulates the binding of ATRIP to ssDNA. The binding of ATRIP to RPA-coated ssDNA enables the ATR-ATRIP complex to associate with DNA and stimulates phosphorylation of the Rad17 protein that is bound to DNA. We also showed this to be true for the budding yeast ortholog of ATRIP, Ddc2. Our data suggest that RPA-coated ssDNA is the critical structure at sites of DNA damage that recruits the ATR-ATRIP complex and facilitates its recognition of substrates for phosphorylation and the initiation of checkpoint signaling.
There is a second branch to DNA damage signaling in mammals that is independent of ATR, the Rad17-911(Rad9, Rad1, Hus1) branch. Rad17 resembles RFC1 and exists in a complex with four RFC subunits, RFC2-5. This homology was originally detected by Tony Carr and led to a hypothesis that the Rad17 complex acts like RFC, which normally loads PCNA onto DNA, but instead loads the PCNA-related 911 complex onto DNA in response to DNA damage. We showed that the human Rad17 protein recruits the 911 complex onto chromatin after damage, strongly supporting the model (146). We found that the chromatin associations of Rad17 and ATR are largely independent, which suggests that they localize to DNA damage independently, also shown in yeast by David Tocyzski. Furthermore, the phosphorylation of Rad17 requires Hus1, suggesting that the Rad1-Rad9-Hus1 complex recruited by Rad17 enables ATR to recognize its substrates. However a central unanswered question remained as to how Rad17 was recruited to chromatin and how it became activated to load the 911 complex onto DNA.
Our previous studies on ATR activation suggested that DNA damage or stalled replication forks generate stretches of RPA-coated ssDNA and this could be a common signal that is sensed to set in motion the DNA damage response signaling. Following that line of reasoning, we found that replication protein A (RPA) stimulates the binding of the Rad17-Rfc2-5 complex to single-stranded DNA (ssDNA), primed ssDNA, and a gapped DNA structure (169). Furthermore, RPA facilitated the recruitment of the Rad9-Rad1-Hus1 complex by the Rad17-Rfc2-5 complex to primed and gapped DNA structures in vitro. These findings suggested that RPA-coated ssDNA is an important part of the structures recognized by the Rad17-Rfc2-5 complex. Unlike replication factor C (RFC), which uses the 3′ primer/template junction to recruit proliferating cell nuclear antigen (PCNA), the Rad17-Rfc2-5 complex can use both the 5′ and the 3′ primer/template junctions to recruit the Rad9-Rad1-Hus1 complex, and it shows a preference for gapped DNA structures. These results generated a model in which the presence of RPA-coated ssDNA sets in motion the independent recruitment of two separate sensors of DNA damage, the ATR/ATRIP and Rad17-Rfc2-5 complexes. The Rad17 complex is activated by a ssDNA-dDNA junction to load 911 onto DNA. Both complexes colocalize to sites of damage and interact providing a failsafe mechanism to initiate signaling. Later work from the Dunphy lab showed that a third component is recruited to these sites by 911, TopBP1, which can be phosphorylated by ATR and which further activates ATR kinase activity to amplify the signal at the site of DNA damage.
Homologous recombination is required for prolonged cell cycle arrest
The DNA damage response (DDR) is brought about by a protein kinase cascade that orchestrates DNA repair through transcriptional and post-translational mechanisms. Cell cycle arrest is a hallmark of the DDR. We screened for cells that lacked damage-induced cell cycle arrest and uncovered a critical role for Fanconi anemia and homologous recombination proteins such as BRCA2 and PALB2 in ATR (ataxia telangiectasia and Rad3-related) signaling. While the checkpoint defect in the FA pathway could be suppressed by turning off NHEJ. Cells lacking BRCA2 could not be suppressed and this suggests that the process of homologous recombination could be prolonging DDR signaling.
Identification of RHINO, a bridge between the 9-1-1 complex and TopBP1
Among the proteins identified in our prolonged checkpoint arrest screen were three DDR candidates, the RNA processing protein INTS7, the circadian transcription factor CLOCK, and a previously uncharacterized protein RHINO, that were recruited to sites of DNA damage. RHINO independently bound the Rad9-Rad1-Hus1 complex (9-1-1) and the ATR activator TopBP1. RHINO was recruited to sites of DNA damage by the 9-1-1 complex to promote Chk1 activation. We suggest that RHINO functions together with the 9-1-1 complex and TopBP1 to fully activate ATR.
Identification of MDC1 – a master regulator of IR induced foci formation
In a search for BRCT-repeat proteins involved in the DNA damage response we identified a novel BRCA1 carboxy-terminal(BRCT) and forkhead-associated (FHA) domain-containing protein we named MDC1 (mediator of DNA damage checkpoint 1), which works with H2AX to promote recruitment of DNA repair proteins to the sites of DNA breaks and which, in addition, controls damage-induced cell-cycle arrest checkpoints (160). MDC1 forms foci that co-localize extensively with g-H2AX foci within minutes after exposure to ionizing radiation. H2AX is required for MDC1 foci formation, and MDC1 forms complexes with phosphorylated H2AX. Furthermore, this interaction is phosphorylation dependent as peptides containing the phosphorylated site on H2AX directly bind MDC1 in a phosphorylation-dependent manner. We showed using siRNA that cells lacking MDC1 are sensitive to ionizing radiation, and that MDC1 controls the formation of damage-induced 53BP1, BRCA1 and MRN foci, in part by promoting efficient H2AX phosphorylation by ATM. In addition, cells lacking MDC1 also fail to activate the intra-S phase and G2/M phase cell-cycle checkpoints properly after exposure to ionizing radiation, which was associated with an inability to regulate Chk1 properly. MDC1 appears to be a primary regulator of DNA damage induced foci formation that amplifies the H2AX signal initiated by ATM and ATR.
Discovery of RIDDLE immunodeficiency syndrome defective in 53BP1-mediated DNA damage signaling – the RNF168 ubiquitin ligase
Cellular DNA double-strand break-repair pathways have evolved to protect the integrity of the genome from a continual barrage of potentially detrimental insults. Inherited mutations in genes that control this process result in an inability to properly repair DNA damage, ultimately leading to developmental defects and also cancer predisposition. We identified a patient with a previously undescribed syndrome, which we have termed RIDDLE syndrome (radiosensitivity, immunodeficiency, dysmorphic features and learning difficulties), whose cells lack an ability to recruit 53BP1 to sites of DNA double-strand breaks (242). As a consequence, cells derived from this patient exhibit a hypersensitivity to ionizing radiation, cell cycle checkpoint abnormalities, and impaired end-joining in the recombined switch regions. Sequencing of TP53BP1 and other genes known to regulate ionizing radiation-induced 53BP1 foci formation in this patient failed to detect any mutations. Therefore, these data indicate the existence of a DNA double- strand break-repair protein that functions upstream of 53BP1 and contributes to the normal development of the human immune system.
The gene for this syndrome was identified by Dan Durocher’s group in a genetic screen and found to be an ubiquitin ligase RNF168, which works with RNF8 to help assemble foci through Mdc1 and ubiquitination of histones.
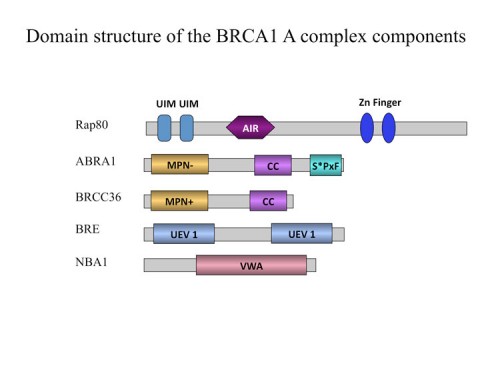
Identification of the BRCA1 A complex and the RNF8/UBC13 ubiquitin ligase that is required for BRCA1 foci formation
As shown by Michael Yaffe and Junjie Chen, the BRCT repeats of BRCA1 form a phospho-peptide recognition motif and are essential for tumor suppression. We used the BRCT repeats of BRCA1 as an affinity reagent to purify peptides that specifically bind BRCA1. This phospho-peptide affinity proteomic analysis identified a protein, Abraxas (202), that we also found as an ATM substrate (201) which directly binds the BRCA1 BRCT repeats through a phospho-SXXF motif where S is serine and F is phenylalanine. Abraxas binds BRCA1 to the mutual exclusion of BACH1 and CTIP, forming a third type of Brca1 complex. Abraxas recruits the ubiquitin-interacting motif (UIM)-containing protein Rap80 to BRCA. Both Abraxas and Rap80 were required for DNA damage resistance, G2-M checkpoint control and DNA repair. Rap80 was required for optimal accumulation of Brca1 on damaged DNA (foci) in response to ionizing radiation and the UIM domains alone were capable of foci formation which implicated the recognition of polyubiquitinated proteins in BRCA1 localization.
Work from David Livingston’s lab, who identified Rap80 independently also found it binds to K63 poly ubiquitin chains but not K48 linked chains. We followed this clue to identify the ubiquitin ligase responsible for generating these chains. Currently, the only E2 known to produce K63-linked polyubiquitin chains is UBC13. A two-hybrid screen with UBC13 was shown to bind 4 different ubiquitin ligases, which we screened for roles in BRCA1 localization. We identified an FHA domain RING finger E3 ubiquitin ligase, RNF8 that binds the UBC13 E2 conjugating enzyme known to form K63-polyubiquitin chains and showed that each are required to recruit the Brca1 A complex to sites of DNA damage (211). Rnf8 localizes to sites of DNA damage through an FHA-domain-containing region which was shown by others to bind phosphorylated Mdc1. We found that Rap80 contains an Abraxas interaction domain, AIR, required for association of Rap80 with Abraxas, Brca1 and Brcc36. Abraxas and Brcc36 associate through coiled-coil domains on each protein. These data suggest a model through which Ubc13 and Rnf8 are recruited to sites of DNA damage through DNA damage-induced phosphorylation of a chromatin associated protein, Mdc1, and generate polyubiquitin chains which then recruit Rap80 and the entire Brca1 A complex to DNA damage foci. This sequential E3 ubiquitin ligase recruitment constitutes a ubiquitin ligase cascade required for DNA repair and checkpoint signaling.
In the process of performing a genetic screen, we identified a gene, NBA1 (new component of the BRCA1 A complex) that is required for resistance to ionizing radiation (226). The NBA1 protein localizes to sites of DNA damage and is required for G2/M checkpoint control. Proteomic analysis revealed that NBA1 is a component of the BRCA1 A complex, which also contains Brca1/Bard1, Abra1, RAP80, BRCC36, and BRE. NBA1 is required to maintain BRE and Abra1 abundance and for the recruitment of BRCA1 to sites of DNA damage. In depth bioinformatics analysis revealed that the BRCA1 A complex bears striking similarities to the 19S proteasome complex. Furthermore, we show that four members of the BRCA1-A complex possess a polyubiquitin chain binding capability, thus forming a complex that might facilitate the deubiquitinating activity of the deubiquitination enzyme BRCC36 or the E3 ligase activity of the BRCA1/BARD1 ligase. These findings provide a new perspective from which to view the BRCA1 A complex.
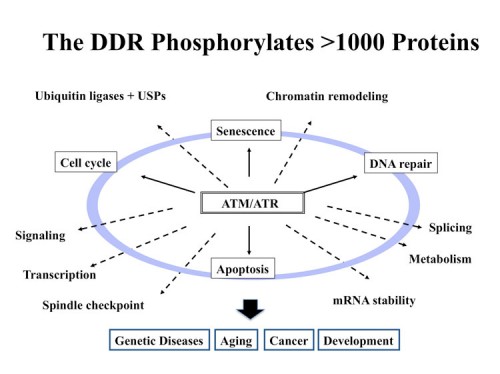
The DNA Damage Phosphoproteome: A new world of DDR effectors
The DNA Damage Response (DDR) is a signal transduction pathway that responds to DNA damage and DNA replication blocks. The DDR is a protein kinase cascade, including ATM, ATR, Chk1 and Chk2. The outlines of the signal transduction portion of this pathway are known, but very little was known about the physiological scope of the DDR. To understand the physiological significance of the DDR pathway, it is imperative that we know the identity of the substrates of these kinases. Thus, we performed a large-scale proteomic analysis of proteins phosphorylated in response to DNA damage on consensus sites recognized by ATM and ATR using an observation that the phospho-specific antibodies we and others had generated to ATM sites, were not as specific as originally thought and cross-reacted with many similar phospho-epitopes. By combining quantitiative mass spectrometry methods known as SILAC with peptide immuoprecipitation, we identified over 900 regulated phosphorylation sites encompassing over 700 proteins (200). Functional analysis of a subset of this dataset indicated that this list is highly enriched for proteins involved in the DNA damage response (DDR). This set of proteins is highly interconnected and identified a large number of protein modules and networks not previously linked to the DDR such as splicing, chromatin remodeling, the ubiquitin system and many repair pathways not already known to be orchestrated by the DDR. This database paints a much broader landscape for the DDR than was previously appreciated and opens new avenues of investigation into the responses to DNA damage in mammals. This database alone has led to the discovery of multiple new DDR proteins including FANCI (197), SLX4 (230), Abraxas (201) and SMARCAL1 (231). We routinely filter genetic screens through this database to identify high priority targets for study.
We have recently performed a quantitative phospho-proteomics screen using SILAC with human, mouse, S. cerevisiae and S.pombe using standard strong cation exchange and IMAC or TiO2 phospho-peptide affinity reagents and now have a comprehensive phospho-proteomics profile for multiple DNA damaging treatrments in these organisms.
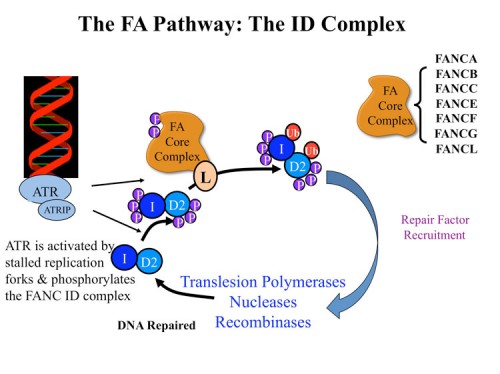
Studies on Fanconi anemia: The Fanconi anemia ID complex
Fanconi anemia (FA) is a developmental and cancer-predisposition syndrome caused by mutations in genes controlling crosslink repair. Several FA proteins form a ubiquitin ligase that controls monoubiquitination of the FANCD2 protein in an ATR-dependent manner. Through analysis of proteins phosphorylated by ATM and ATR in response to DNA damage, we identified a subset of proteins that are required for resistance to DNA crosslinking agents such as mitomycin C, MMC. One such protein has sequence relatedness with the Fanconi anemia protein FANC D2 and through genetic analyses we determined it encoded a previously unidentified component of the Fanconi anemia pathway, FANCI (197). FANCI is highly conserved, and shares sequence similarity with FANCD2, likely evolving from a common ancestral gene. The FANCI protein associates with FANCD2 and together, as an ID complex, they load onto chromatin in response to DNA damage. Like FANCD2, FANCI is monoubiquitinated on a lysine critical for its function. Unexpectedly, ubiquitination of each protein is essential for the maintenance of ubiquitin on the other, indicating the existence of a dual ubiquitin locking mechanism required for the ability of the FANCI-FANCD2 (ID) complex to localize to DNA damage foci and regulate crosslink repair. Mutation in the FANCI gene is responsible for loss of a functional FA pathway in a patient with Fanconi anemia complementation group I.
We initiated a collaboration with Minoru Takata’s group to examine the FANCI gene in the chicken DT40 cell system (221). We found that FANCI null mutants had a phenotype indistinguishable from FANC D2 mutants. We analyzed the significance of ATM/ATR phosphorylation on FANC ID complex my mutating SQ and TQ sites. Removal of all such sites from FANC D2 had no effect on FANC D2 activity. Multiple alanine substitution mutations in six conserved and clustered Ser/Thr-Gln motifs of FANCI largely abrogate monoubiquitination and focus formation of both FANCI and FANCD2, resulting in loss of DNA repair function. Conversely, FANCI carrying phosphomimic mutations on the same six residues induces constitutive monoubiquitination and focus formation of FANCI and FANCD2, and protects against cell killing and chromosome breakage by DNA interstrand cross-linking agents. We propose that the multiple phosphorylation of FANCI serves as a molecular switch in activation of the Fanconi anemia pathway. Mutational analysis of putative phosphorylation sites in human FANCI indicates that this switch is evolutionarily conserved. We think that FANCI phosphorylation allows the ID complex to be recognized by the FANC ubiquitin ligase core complex and that ubiquitination activates the complex and locks it onto chromatin at the sites of damage where the ATR kinase is located.
To explore the biochemical role of the FANC ID complex we cloned the Xenopus FANCI gene and collaborated with Johannes Walter’s group who had established a Xenopus extract in vitro crosslink repair assay. Using this cell-free system we depleted the ID complex and showed that the FANCI-FANCD2 complex is required for replication-dependent ICL repair (232). Removal of the ID complex from extracts inhibits nucleolytic incisions near the ICL as well as translesion DNA synthesis past the lesion. Reversal of these defects requires ubiquitylated FANCI-FANCD2. Thus, the FANC ID complex is required for very early events in the repair of crosslinks.
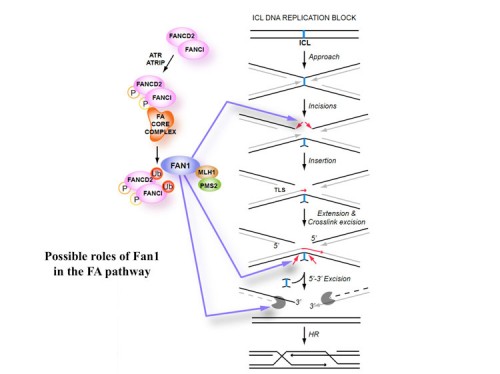
To identify the mechanism through which ubiquitylated ID complex directs repair, we performed an shRNA screen to identify genes required for mitomycin C (MMC) resistance. We identified two new nucleases, EXDL2 and FAN1. FAN1 contains a ubiquitin-binding domain, UBZ, a SAP, TPR and nuclease domains. We found FAN1 co-localizes at sites of DNA damage with the ID complex dependent on FAN1’s ubiquitin binding domain, the ID complex and monoubiquitination of FANCD2 (240). FAN1 possesses an intrinsic endonuclease activity cleaving nicked and branched structures, as well as 5’-3’ exonuclease activity. Thus, FAN1 is a repair nuclease that is recruited to sites of crosslink damage in part through binding the ubiquitinated ID complex through its UBZ domain and could participate in one of the four incisions required to remove a interstrand crosslink or could prepare the cleaved strands for subsequent homologous recombination by resecting the ends to leave the 3’ overhangs needed to initiate homologous recombination.
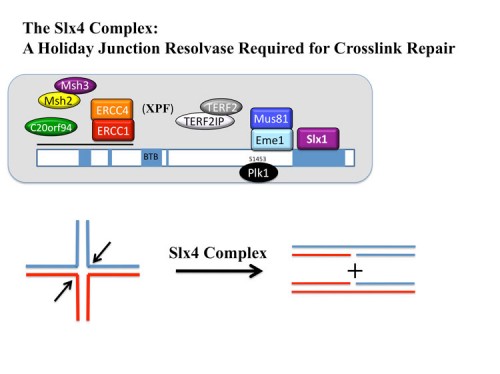
Identification of SLX4, a molecular toolkit for resolution of Holiday Junctions
Analysis of our collection of ATM and ATR substrates for genetic phenotypes identified a BTB domain protein, BTBD12, which we showed was required for resistance to a number of different DNA damaging agents. Proteomic and sequence analyses with Wade Harper’s lab led to the discovery that BTBD12 was the human ortholog of the budding yeast DNA repair factor Slx4p and D. melanogaster MUS312 (230). We found that human SLX4 forms a multi-protein complex with the ERCC4(XPF)-ERCC1, MUS81-EME1, and SLX1 endonucleases and also associates with MSH2/MSH3 mismatch repair complex, telomere binding complex TERF2(TRF2)-TERF2IP( RAP1), the protein kinase PLK1 and the uncharacterized protein C20orf94. Depletion of SLX4 causes sensitivity to mitomycin C and camptothecin and reduces the efficiency of DSB repair in vivo. SLX4 complexes cleave 3’ flap, 5’ flap, and replication fork structures; yet unlike other endonucleases associated with SLX4, the SLX1-SLX4 module promotes symmetrical cleavage of static and migrating Holliday junctions (HJs), identifying SLX1-SLX4 as a HJ resolvase. Thus, SLX4 assembles a modular toolkit for repair of specific types of DNA lesions and is critical for cellular responses to replication fork failure.
The crystal Structure of the Fanconi Amenia ID Complex
Through acollaboration with Nicola Paveltich at Memorial Sloan Kettering, the structure of the FANC ID complex was determined. The 3.4 angstrom crystal structure of the ~300 kilodalton ID complex reveals that monoubiquitination and regulatory phosphorylation sites map to the I-D interface, suggesting that they occur on monomeric proteins or an opened-up complex and that they may serve to stabilize I-D heterodimerization. The 7.8 angstrom electron-density map of FANCI-DNA crystals and in vitro data show that each protein has binding sites for both single- and double-stranded DNA, suggesting that the ID complex recognizes DNA structures that result from the encounter of replication forks with an ICL. We believe that the localization to the sDNA-dsDNA structures at stalled replication forks positions the ID complex to recruit nucleases such as FAN1 and recombinases such as the SLX4 resolvase complexes to the sites where repair is needed.
Deubiquitination in the DDR: USP28 controls the stability of multiple DDR proteins
The ATM-53BP1-Chk2-p53-PUMA pathway is a major regulator of DNA damage induced apoptosis in response to double-strand breaks in vivo. Through analysis of 53BP1 complexes we discovered a new ubiquitin protease, USP28, which regulates this pathway (193). Using a human cell line that faithfully recapitulated the ATM-53BP1-Chk2-p53-PUMA pathway, we show that USP28 is required to stabilize Chk2 and 53BP1 in response to DNA damage. In this cell line, both USP28 and Chk2 are required for DNA damage induced apoptosis and they accomplish this in part through regulation of the p53 induction of proapoptotic genes like PUMA. A number of other proteins in the DNA damage checkpoint pathway including several mediators such as Mdc1, Claspin, and TopBP1 are also regulated by USP28. Interestingly, in the absence of USP28, DNA damage causes the appearance of a shorter form of p53. Our studies implicate DNA damage induced ubiquitination and deubiquitination as a major regulator of the DNA damage response. A current question concerns why loss of USP28 causes these phenotypes only in some cell lines.
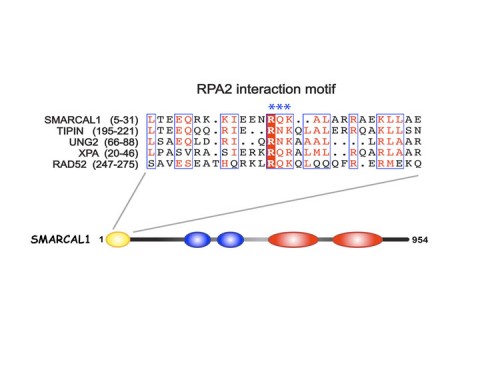
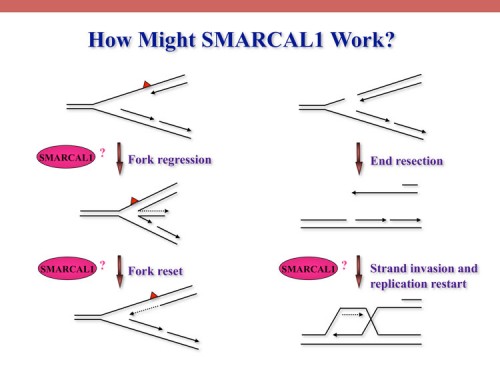
SMARCAL1, mutated in Schimke immuno-osseous dysplasia (SIOD), controls replication fork stability implicating replication stress as the cause of SIOD
The integrity of genomic DNA is continuously challenged by the presence of DNA base lesions or DNA strand breaks. Using proteomic screening we identified a new DNA damage response protein, SMARCAL1, which is a member of the SNF2 family and is mutated in Schimke immuno-osseous dysplasia (SIOD) (231). We demonstrate that SMARCAL1 directly interacts with RPA and is recruited to sites of DNA damage in an RPA-dependent manner.
SMARCAL1-depleted cells display sensitivity to DNA damaging agents that induce replication fork collapse and exhibit slower fork recovery and delayed entry into mitosis following S phase arrest.
Furthermore, SIOD patient fibroblasts reconstituted with SMARCAL1 exhibit faster cell cycle progression after S-phase arrest. Thus, the symptoms of SIOD may be caused, at least in part, by defects in the cellular response to DNA replication stress.

Tipin and Timeless and a Role for Circadian Rhythms in the DDR
Tipin is a mammalian protein that interacts with Timeless, which plays a role in DNA damage checkpoint responses. We found that Tipin is a nuclear protein that associates with the replicative helicase, the MCM proteins, and protects cells against genotoxic agents (194). Tipin is required for efficient cell cycle arrest in response to DNA damage, and depletion of Tipin renders cells sensitive to ionizing radiation as well as replication stress. Loss of Tipin results in spontaneous gamma-H2AX foci, a marker for DNA double strand breaks. We find that Tipin and Timeless form a complex that maintains the level of both proteins in cells, and that loss of either one will lead to the loss of the interacting partner. This observation explains the similar checkpoint phenotypes observed in both Tipin and Timeless depleted cells.
In yeast these proteins promote stable replication fork maintenance at pause sites. In addition, several proteins such as Timeless, Per2, Cry and CLOCK appear to have roles in both the DNA replication stress response and in circadian rhythms. Furthermore, mice lacking Per2 are tumor prone. An open question is how and why these two systems are coupled. One idea is that circadian rhythms evolved in algae in the ocean that needed to sense light and temperature in order to optimize their metabolic success. They evolved to sense UV light as a time measuring mechanism and DNA damage was occurring simultaneously with the sensing of UV light by chryptochromes. Perhaps these systems co-evolved to have common functions very early on.
Polycomb Complexes and DNA damage
Many proteins that respond to DNA damage are characterized by recruitment to DNA lesions. We employed a proteomics approach that coupled isotopic labeling with chromatin fractionation and mass spectrometry to uncover proteins that associate with damaged DNA. We found that multiple polycomb group protein family members are recruited to DNA lesions following UV laser micro-irradiation. This recruitment requires PARP activity, but not ATM, ATR, or histone H2AX. We found that loss of polycomb repressive complex 2 (PRC2) components results in sensitivity of mammalian cells and, through a collaboration with Monica Colaiacovo’s lab, sensitivity of C. elegans embryos to ionizing radiation suggesting that repressive chromatin modification at sites of DNA damage may be important for DNA repair.
DNA damage induced protein modification
The cellular machinery of the DNA damage response (DDR) consists of a dynamic network of multiprotein complexes whose hierarchical assembly and subsequent downstream signaling rely upon an array of posttranslational modifications, with phosphorylation and ubiquitination playing particularly prominent roles. Using a proteomic approach involving mass spectrometry and SILAC (stable isotope labeling of amino acids in cell culture), we are identifying such novel post-translational modifications induced by radiation and other forms of DNA damage. This work will complement our previous study identifying more than 900 IR-regulated ATM and ATR phosphorylation sites, an effort which significantly expanded the known physiological scope of the DDR.
Homologous recombination
Genomic stability is essential to the survival of all organisms, and to safeguard genetic information from various DNA lesions, conserved mechanisms of DNA damage response have evolved. Homologous recombination (HR) is an error-free, replication-dependent mechanism of DNA repair that allows re-synthesis of damaged DNA from an intact homologous template. Our current understanding of the protein networks that regulate HR in mammals is comprehensive; however, many mediators of this repair mechanism remain unidentified. To identify novel HR factors, we have conducted a genome-wide, RNAi-based screen in a human osteosarcoma cell line. We are now examining the candidates from this screen to gain a better understanding of the various cellular mechanisms that coordinate HR.
Genetic screens for resistance to DNA damaging agents
A better grasp of how the process of sensing and repairing DNA damage works goes hand in hand with a better understanding of cancer and other diseases related to cell growth. Our lab in collaboration with Dr. Greg Hannon’s lab (Cold Spring Harbor) has constructed a large-scale library of RNAi-inducing shRNA expression vectors targeting human and mouse genes and the use of these libraries for screens to identify novel tumor suppressors (181) and genes involved in cancer cell proliferation was recently demonstrated (205). We are using these shRNA libraries along with microarray deconvolution technology to identify novel players in the mammalian DNA damage response and in particular genes whose knockdown leads to cellular sensitivity or resistance to ionizing radiation and DNA crosslinking agents such as mitomycin C, MMC.
Wolf-Hirschhorn syndrome is due to a defect in the DNA replication response
Wolf-Hirschhorn syndrome (WHS) is a malformation syndrome associated with growth retardation, mental retardation, and immunodeficiency resulting from a hemizygous deletion of the short arm of chromosome 4, called the WHS critical region (WHSC). The WHSC1 gene is located in this region, and its loss is believed to be responsible for a number of WHS characteristics. We identified WHSC1 in a genetic screen for genes involved in responding to replication stress, linking Wolf-Hirschhorn syndrome to the DNA damage response (DDR). Here, we report that the WHSC1 protein is a member of the DDR pathway. WHSC1 localizes to sites of DNA damage and replication stress and is required for resistance to many DNA-damaging and replication stress-inducing agents. Through its SET domain, WHSC1 regulates the methylation status of the histone H4 K20 residue and is required for the recruitment of 53BP1 to sites of DNA damage. We propose that Wolf-Hirschhorn syndrome results from a defect in the DDR.
Summary of the Yeast DDR and RNR Regulation
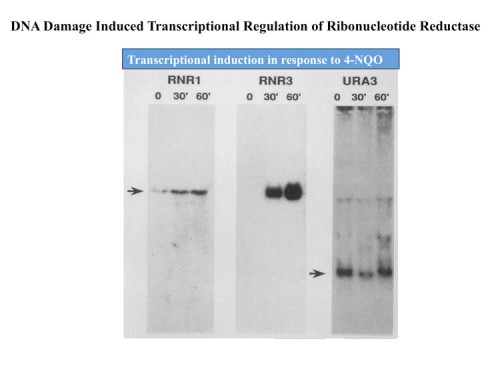
Identification of RNR DNA damage inducibility
Early work from the Elledge lab sprang from the chance discovery that the small subunit of the enzyme ribonucleotide reductase, RNR2, was under stringent transcription control in response to DNA damage and DNA replication stress (15, 19, 20). RNR catalyzes the conversion of ribonucleotides to deoxyribonucleotides needed for DNA synthesis, and thus sits at the crux of a very critical pathway. Further work identified additional subunits of the enzyme, RNR1, 3 and 4 and all four subunits were highly inducible as well (87, 22).
Elucidation of the DNA damage response pathway in S. cerevisiae
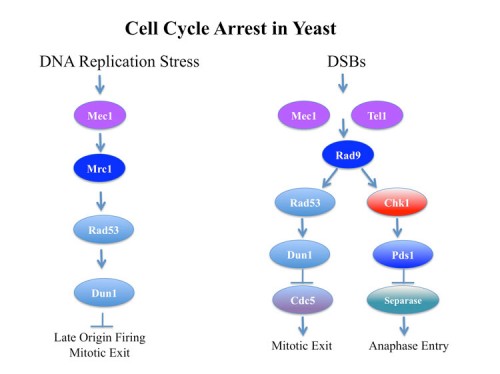
The inducibility of the RNR genes suggested the existence of a regulatory pathway that could sense and respond to DNA damage and replication stress. Using the promoters of the RNR genes as tools to dissect this pathway, genetic screens were carried out to identify constitutive (29) and uninducible mutants (38) altered in the ability to send these signals. Through a large number of genetic screens of many types coupled with molecular biological analyses, the outlines of the yeast DNA damage response pathway emerged in which two PI3-related kinases, Tel1(yeast ATM) and Mec1/Sad3(Yeast ATR) (73), sense DNA damage and replication blocks and then activate a family of checkpoint kinases Chk1 (116) and Rad53/Sad1/Spk1(yeast Chk2) (49). Rad53 also regulates the Dun1 kinase (also a Chk2 family kinase) making an extensive protein kinase cascade. The response to replication blocks were transduced through a Mediator protein Mrc1 (138), which moves along the DNA with the replication fork (165) and becomes highly phosphorylated in response to DNA damage. Once phosphorylated, Mrc1 facilitates the activation of Rad53 by recruiting it to the Mec1 kinases where is phosphorylated and activated. In response to non-replicative damage, the Rad9 mediator protein performs the same function to activate Rad53. We also found mutations in the DNA polymerase epsilon protein, Pol2, that reduced DDR signaling on response to DNA replication stress (58) implying that the DNA damage response signal emanates from the replication fork, a fact we were able to later confirm in mammals.

The mechanism of RNR transcriptional induction
The mechanism of transcriptional induction of the RNR genes is through the Rad53- and Dun1-dependent phosphorylation and inactivation of the Crt1(Constitutive RNR Transcription) (also known as Rfx1) repressor (100). Crt1 is a member of the RFX family of X-box binding transcription factors. It acts by recruiting two other proteins found in the Crt screen, the transcriptional co-repressors Tup1 and Ssn6, to the promoters of the RNR genes.
The mechanism of cell cycle arrest in response to DNA damage
In response to DNA damage, yeast arrest in a metaphase equivalent state prior to anaphase entry and this arrest requires the Mec1-Rad53-Chk1-Dun1 kinases. The mechanism of cell cycle arrest was found to be two-fold: stabilization of Pds1 (yeast securin) through Chk1 phosphorylation (136) and inhibition of maintenance of Bub/Bfa1 GAP activity by inhibition of its activator Cdc5 (yeast polo kinase, Plk1) through Rad53 activity (116). Blocking Cdc5 prevents inhibition of the GAP activity of the Bub2/Bfa1 (143) that normally occurs to promote Tem1 activation, a G-protein involved in anaphase initiation.
The essential function of Mec1 and Rad53 involve regulation of DNA replication
Both Mec1 and Rad53 are essential genes that are required for cell cycle progression in every cell cycle. We found that the essential functions of Mec1 and Rad53 can be suppressed by increasing the amount or activity of ribonucleotide reductase. Our lab found that Mec1 and Rad53 play a critical role in the recovery from DNA replication blocks (101). Even a transient inhibition of DNA replication prevents the complete replication of chromosomes in these mutants.
Post-transcriptional regulation of ribonucleotide reductase
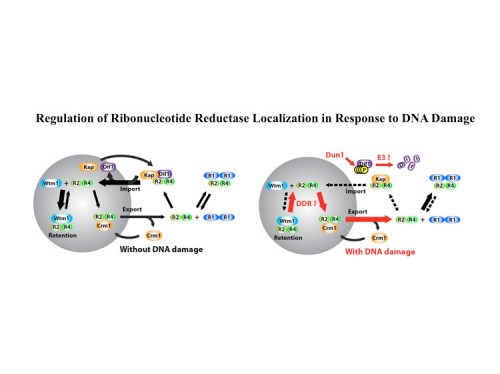
In addition to transcriptional regulation of Rnr expression, the DNA damage response pathway activates Rnr function by two separate mechanisms. One is by control of localization of the Rnr2 protein in the nucleus while the large subunit, Rnr1, localizes to the cytoplasm. This localization was first discovered by Tony Carr in S. pombe and Mingxia Huang in S. cerevisiae. During S phase or in the presence of DNA damage, the Rnr2 leaves the nucleus and joins Rnr1 in the cytoplasm to make dNTPs for DNA synthesis. This is accomplished through the activity of two proteins Wtm1 and Dif1. Wtm1 acts as a nuclear anchor that binds Rnr2 and sequesters it in the nucleus (188). Dif1 is a nuclear importer that binds Rnr2 and imports it into the nucleus. In response to DNA damage or replication stress, Dif1 is phosphorylated and inactivated for binding and transport of Rnr2 and is targeted for destruction through an unknown ubiquitin ligase pathway, resulting in the cytoplasmic localization of Rnr2 (216). It is likely that Rnr2 binding to Wtm1 is also regulated, although how that occurs is presently unknown. A second pathway of Rnr regulation is through the Sml1 protein, found by Rodney Rothstein, which binds and inhibits Rnr1 in the cytoplasm. Sml1 is phosphorylated by Dun1 and targeted for degradation. Thus multiple mechanisms act to negatively regulate Rnr and inactivation of any one of them, Crt1, Dif1, Wtm1 or Sml1 can suppress the lethality of Mec1 and Rad53 mutations.
The Mammalian DNA Damage Response
Chk1, a central DNA damage signal tranducer is an essential gene and a haplo-insufficient tumor suppressor
We previously cloned human Chk1, an evolutionarily conserved protein kinase, which we implicated in cell cycle checkpoint control through phosphorylation of Cdc25 molecules (88). By gene disruption, we showed that CHK1 deficiency results in a severe proliferation defect and death in embryonic stem (ES) cells, and peri-implantation embryonic lethality in mice (125). Through analysis of a conditional CHK1-deficient cell line, we found that ES cells lacking Chk1 have a defective G(2)/M DNA damage checkpoint in response to gamma-irradiation (IR). CHK1 heterozygosity enhances the tumorigenesis phenotype of WNT-1 transgenic mice. We found that Chk1 is phosphorylated on serine 345 (S345) in response to UV, IR, and hydroxyurea (HU) providing an assay for activation of the Chk1 pathway. Overexpression of wild-type Atr enhances, whereas overexpression of the kinase-defective mutant Atr (or treatment with siRNA to ATR) inhibits S345 phosphorylation of Chk1 induced by UV. These data indicate that Chk1 plays an essential role in the mammalian DNA damage checkpoint, embryonic development, and tumor suppression, and that ATR regulates Chk1.
The early embryonic lethality hampered efforts to look at Chk1 function in adult tissues. Therefore, we made a conditional Chk1 mouse (174). Using these conditional Chk1 animals, we found that proliferating mammary cells lacking Chk1 undergo apoptosis leading to developmental defects. Conditional Chk1 heterozygosity increased the number of S phase cells and caused spontaneous DNA damage. Chk1+/- epithelia also exhibit a miscoordinated cell cycle in which S phase cells display an early mitotic phenotype. These cells maintain high levels of Cdc25A, which can promote inappropriate cell cycle transitions. Thus, Chk1 heterozygosity results in three distinct haploinsufficient phenotypes that can contribute to tumorigenesis: inappropriate S phase entry, accumulation of DNA damage during replication, and failure to restrain mitotic entry.
Chk2, an ATM and ATR regulated kinase that regulates p53
We identified the human Chk2 gene and found that it is activated by ATM phosphorylation in response to DNA damage (104, 127). We generated Chk2-deficient mice by gene targeting with Tak Mak (121). Chk2(-/-) embryonic stem cells failed to maintain gamma-irradiation-induced arrest in the G2 phase of the cell cycle. Chk2(-/-) thymocytes were resistant to DNA damage-induced apoptosis were defective for p53 stabilization and induction of p53-dependent transcripts such as p21 in response to gamma irradiation. Chk2 directly phosphorylates p53 on serine 20 in vitro. It also phosphorylated Cdc25 in vitro.
Like p53, Chk2 is mutated in patients with Li-Fraumeni syndrome. Unlike ATM(-/-) and p53(-/-) mice, Chk2(-/-) mice do not spontaneously develop tumors, although Chk2 does suppress 7,12-dimethylbenzanthracene (DBA)-induced skin tumors. Tissues from Chk2(-/-) mice, including those from the thymus, central nervous system, fibroblasts, epidermis, and hair follicles, show significant defects in IR-induced apoptosis or impaired G(1)/S arrest. Quantitative comparison of the G(1)/S checkpoint, apoptosis, and expression of p53 proteins in Chk2(-/-) versus ATM(-/-) thymocytes suggested that Chk2 can regulate p53-dependent apoptosis in an ATM-independent manner. IR-induced apoptosis was restored in Chk2(-/-) thymocytes by reintroduction of the wild-type Chk2 gene but not by a Chk2 gene in which the sites phosphorylated by ATM and ATR were mutated to alanine.
Thymocytes lacking Chk1 eventually die by a p53-independent apoptosis. However, loss of Chk2 suppresses this lethality indicating that Chk2 has p53-independent functions as well as p53-dependent functions (149).
A link between Microcephaly and the DDR through BRIT1/MCPH1
We originally identified BRIT1 (BRCT-Repeat Inhibitor of hTERT expression) as a repressor of human telomerase (hTERT) expression (161). Further analysis of cells lacking BRIT1 activity by siRNA revealed that BRIT1 acts as a regulator of both the intra-S and G2/M checkpoints (184). When BRIT1 expression is depleted, cells lose IR-induced cell cycle arrest and become IR sensitive. BRIT1 is a chromatin-associated protein that forms irradiation-induced nuclear foci that colocalize with g-H2AX foci. BRIT1 is also required for the expression of both BRCA1 and Chk1 and phosphorylation of Nbs1. Thus, the checkpoint defects in the absence of BRIT1 are likely to result from its regulation of Nbs1, BRCA1 and Chk1. BRIT1 is identical to the newly discovered MCPH1 gene, found mutant in patients with primary microcephaly. The ATR-Chk1 pathway is defective in Seckel Syndrome, another microencephaly disorder. We propose that the microencephaly observed in patients with MCPH1 deficiencies is due to disruption of the ATR-BRCA1-Chk1 signaling pathway also disrupted in Seckel Syndrome patients.
Connections between the DDR and Brca1
Brca1 was shown to undergo phosphorylation in response to DNA damage from David Livinston’s lab. We showed that Brca1 phosphorylation was controlled by ATM in response to ionizing regulation and by ATR in response to replication stress (115). This finding made a connection between two known tumor suppressor pathways, ATM and Brca1. Furthermore, is shored up the somewhat anecdotal links between ATM heterozygousity and breast cancer and begged the question as to whether mammograms of individuals who are heterozygous for ATM are a potential health hazard for the recipients.


