E3 Ubiquitin Ligase Biology
While scientists know a great deal about how cell regulation is controlled at the transcriptional level, very little is known about how the proteome is remodeled through regulated protein stability. Our lab and that of our collaborators discovered the largest family of E3 ubiquitin ligases known as the CRLs, cullin-ring ligases. We have been interested in developing general methods to identify the substrates of these ligases to better understand how signal transduction remodels the proteome. Some of our studies are described below.
- General Ubiquitin Introduction
- Recent Studies
- Older Work
- The discovery of F-box proteins and the SCF family of modular E3 ubiquitin ligases
- The SCF as the founding member of a large set of modular Cullin-based ligases
- The RING domain as an E2 recruitment domain revealed a large family of E3 ligases
- F-box protein gene families in eukaryotes
- Development of the GPS (global protein stability) system for the genome-wide measurement of protein stability
- Identification of Cul1 substrates
General Ubiquitin Introduction
The addition of poly-ubiquitin chains to substrate proteins, and their subsequent destruction by the proteasome, is a process that permeates all aspects of cellular biology. The formation of ubiquitin–protein conjugates requires three components: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a specificity factor (E3) that functions in substrate recognition. Polyubiquitinated proteins with K48 ubiquitin chains are degraded by the 26S proteasome. Other ubiquitin linkages have non-proteosomal functions although there is much to be learned about how each linkage is interpreted by the cell. How and when specific proteins are ubiquitinated is the critical issue concerning a systems level understanding of the proteome, and E3 complexes provide the key to regulated proteolysis.
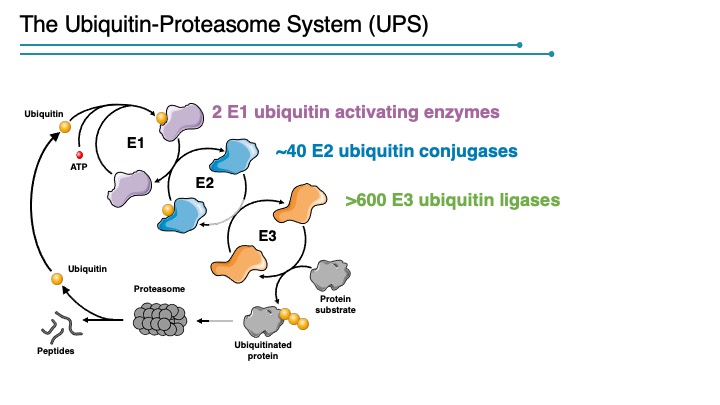
Recent Studies
Ubiquitin control of transcription factors
We have begun an analysis if the stability of transcription factors and their regulation by the UPS pathway with a focus on Cullin-Ring-Ligases. During this study we recognized the striking instability of the TFE3 and MITF master transcription factors that maintain metabolic homoeostasis by regulating lysosomal, melanocytic, and autophagy genes. Previous studies posited that their cytosolic sequestration, mediated by the Rag GTPases-mTORC1 nutrient-sensing pathway, was key for suppressing transcriptional activity in the presence of nutrients. However, using Global Protein Stability (GPS) assays for examining the stability of transcription factors, we found an even more critical role for regulated protein stability in the control of TFE3 and MITF.
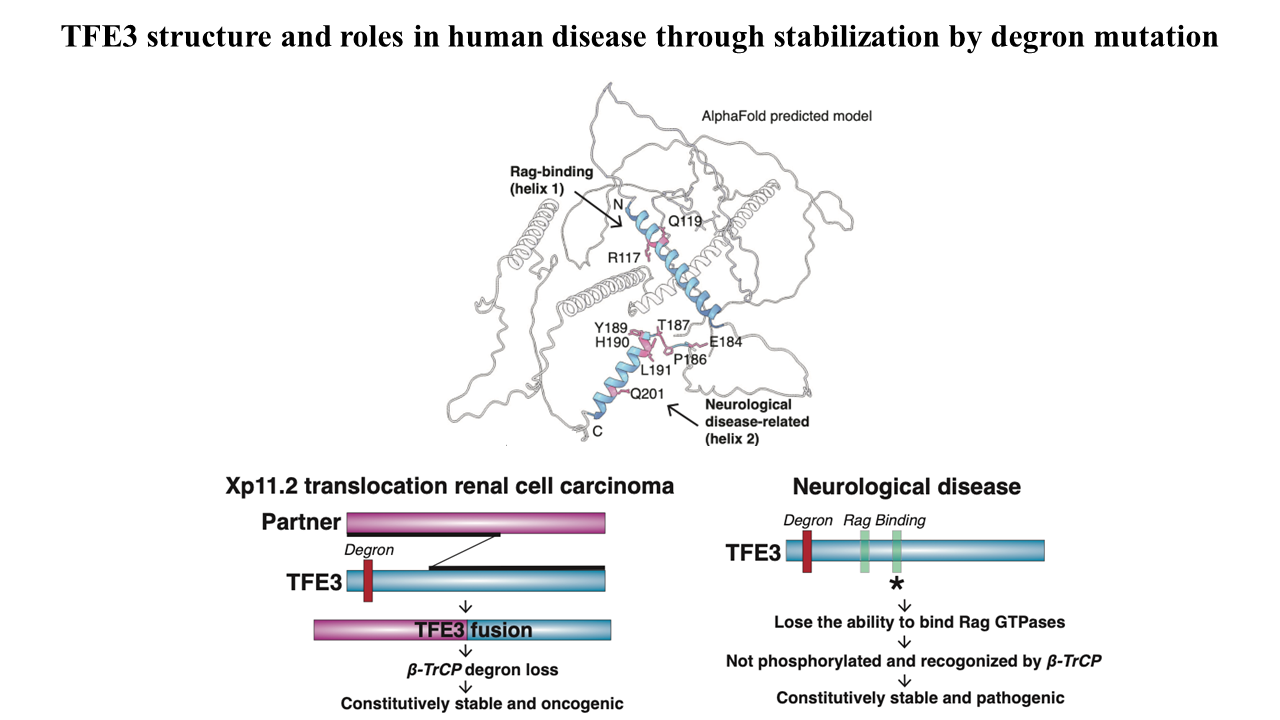
Amino acids promote the recruitment of TFE3 and MITF to the lysosomal surface, activating an evolutionarily conserved phospho-degron leading to their degradation. Elucidation of the minimal functional degron uncovered a conserved alpha helix (helix 2) which we showed to be required for lysosomal surface recruitment and activation of the degron.
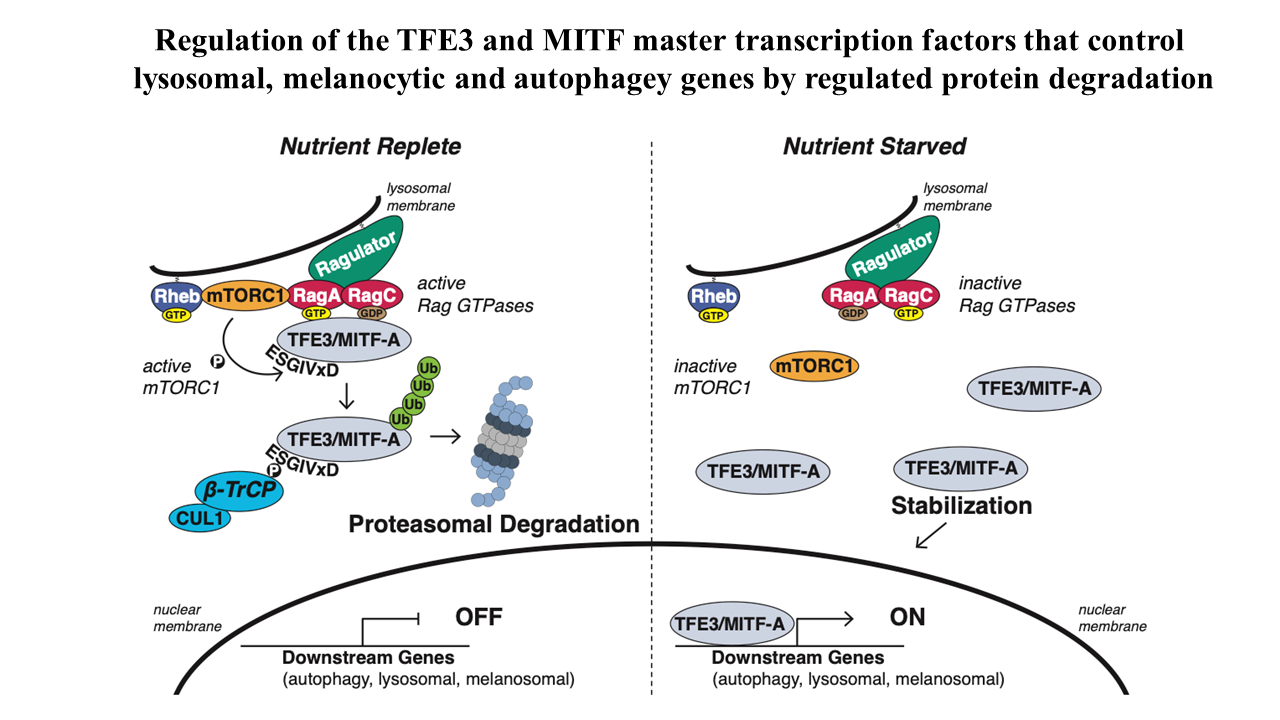
A severe neurodevelopmental syndrome is caused by mutations that cluster in the TFE3 helix 2 and result in the stabilization of TFE3, illuminating the molecular basis for this debilitating disease. Furthermore, translocation renal cell carcinoma is caused by a genomic rearrangement of TFE3, leading to oncogenic TFE3 fusion proteins. These TFE3 fusions recurrently remove the degron and its restoration severely reduces the levels of the fusion oncogenes. Therefore, two divergent pathologies converge on the loss of this protein stability regulation by nutrients.
The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons
Degrons are minimal elements that mediate the interaction of proteins with degradation machineries to promote proteolysis. Despite their central role in proteostasis, the number of known degrons remains small, and a facile technology to characterize them is lacking. Using a strategy combining global protein stability (GPS) profiling with a synthetic human peptidome, we identify thousands of peptides containing degron activity.
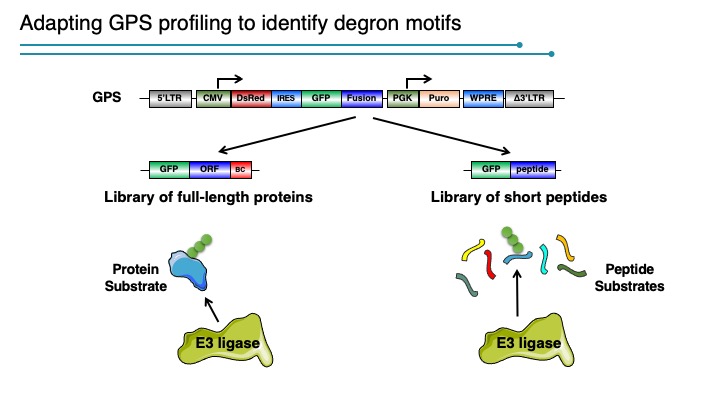
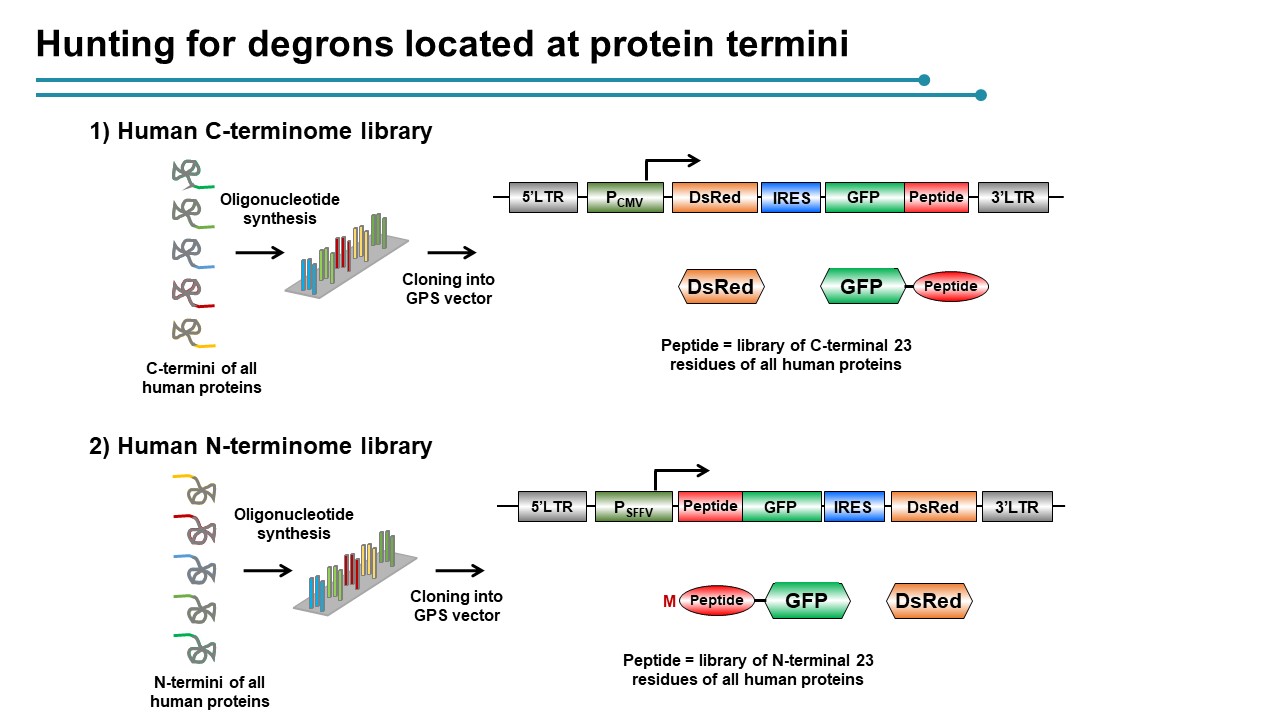
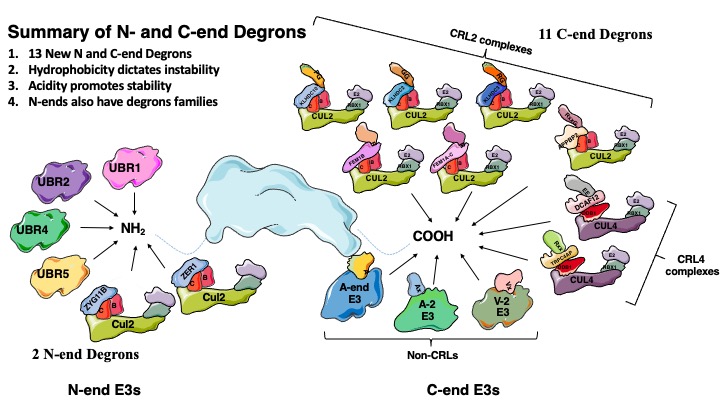
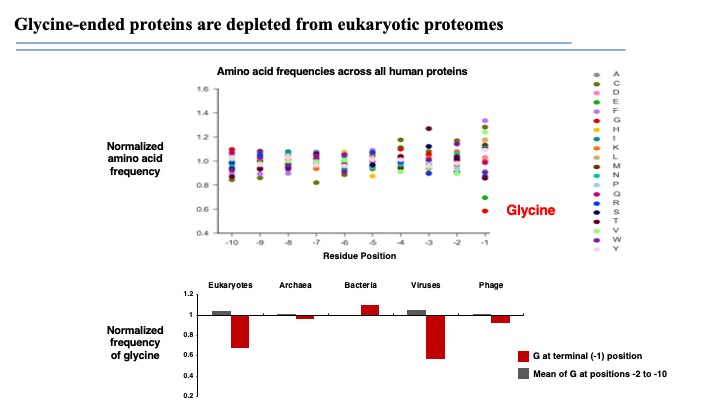
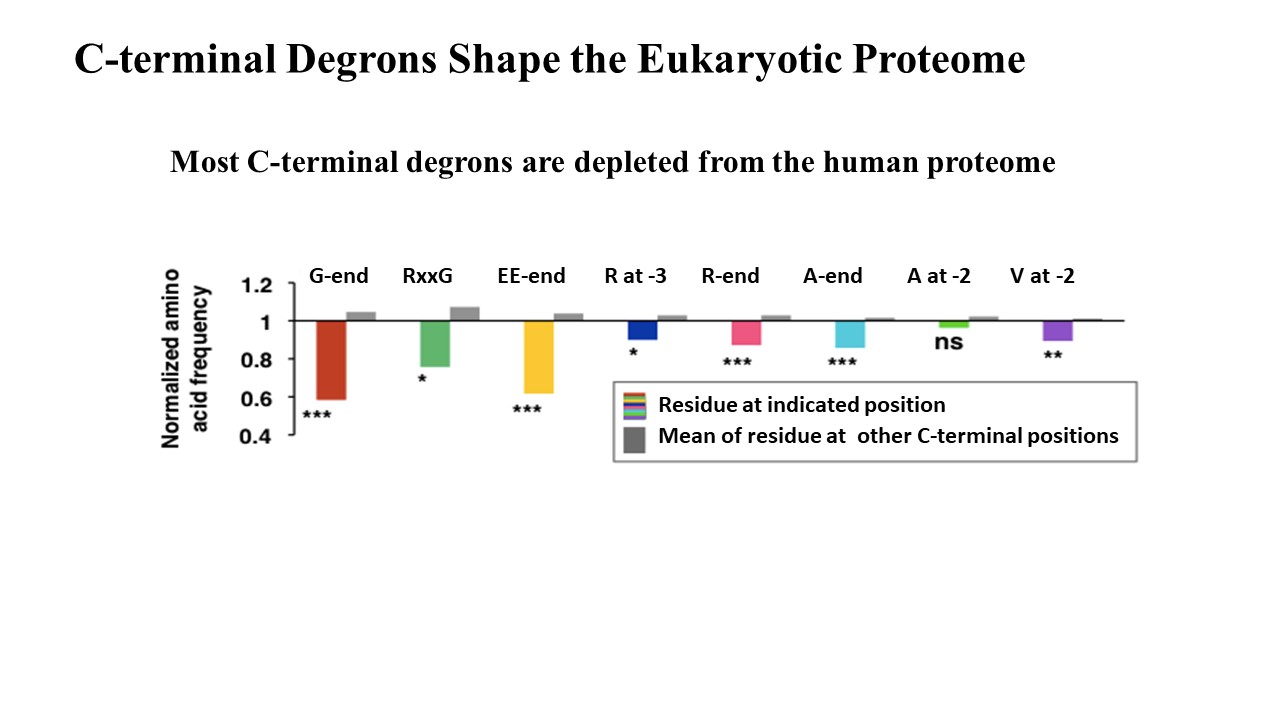
Employing CRISPR screening, we establish that the stability of many proteins is regulated through degrons located at their C terminus. We characterize eight Cullin-RING E3 ubiquitin ligase (CRL) complex adaptors that regulate C-terminal degrons, including six CRL2 and two CRL4 complexes, and computationally implicate multiple non-CRLs in end recognition. Proteome analysis revealed that the C termini of eukaryotic proteins are depleted for C-terminal degrons, suggesting an E3-ligase-dependent modulation of proteome composition. Thus, we propose that a series of “C-end rules” operate to govern protein stability and shape the eukaryotic proteome.
A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation
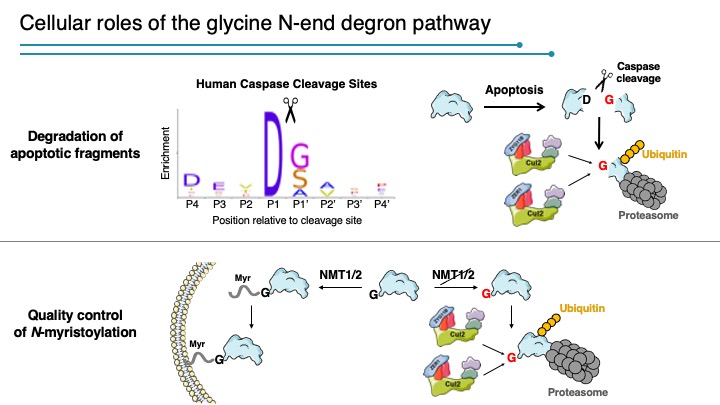
The N-terminal residue influences protein stability through N-degron pathways. We used GPS stability profiling of the human N-terminome to uncover multiple additional features of N-degron pathways. In addition to uncovering extended specificities of UBR E3 ligases, we characterized two related Cullin-RING E3 ligase complexes, Cul2ZYG11B and Cul2ZER1, that act redundantly to target N-terminal glycine. N-terminal glycine degrons are depleted at native N-termini but strongly enriched at caspase cleavage sites, suggesting roles for the substrate adaptors ZYG11B and ZER1 in protein degradation during apoptosis. Furthermore, ZYG11B and ZER1 were found to participate in the quality control of N-myristoylated proteins, in which N-terminal glycine degrons are conditionally exposed after a failure of N-myristoylation. Thus, an additional N-degron pathway specific for glycine regulates the stability of metazoan proteomes.
Older Work
The Discovery of F-box proteins and the SCF family of Modular E3 Ubiquitin Ligases
F-box proteins are modular adaptors that filter the proteome. They are post-translation factors that are analogous to microRNAs. MicroRNAs are post-transcription factors that edit the transcriptome controlling mRNA abundance in the same way F-box proteins edit the proteome controlling protein abundance. Both factors contain sequence identification capability, can recognize multiple targets and plug into an enzymatic machine in a modular fashion to change the substrate specificity of the machine.
The F-box Hypothesis
In 1996 our group published the discovery of a group of proteins called F-box proteins which we linked, together with Skp1, to control of protein stability (77). Through genetic and biochemical analyses we formulated the F-box hypothesis which postulated that F-box proteins acted as substrate adaptors that recruit substrates to a modular ubiquitin ligase.
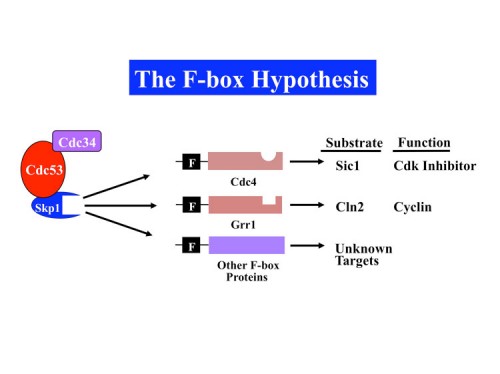
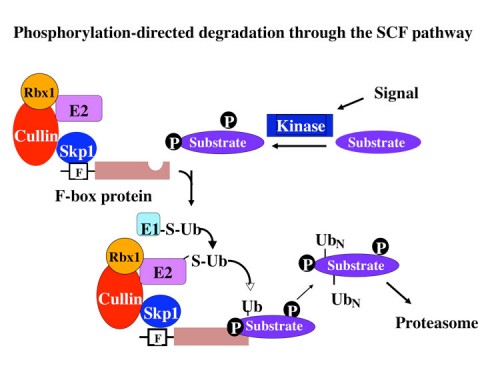
F-box proteins generally contain a minimum of 2 domains; the F-box motif which binds to Skp1 and a scaffolding protein called a Cullin (first implicated in protein stability by Mike Tyers and Mark Goebl), and a protein-protein interaction domain that allows recognition of substrates. Subsequent biochemical analyses carried out with our colleague Wade Harper, and independently by Ray Deshaies, confirmed this hypothesis by reconstituting an active E3 ubiquitin ligase we named the SCF (Skp1-Cullin-F-box) (90). We found that different F-box proteins allowed ubiquitination of different substrates. There are three general classes of F-box proteins, those that contain a WD40 repeat domain, FBXW proteins, those that contain a leucine rich repeat (LRR) domain, and those that do not contain a previously identified protein-interaction domain, FBXO proteins (118). Importantly we found that WD40 and LRR repeat domains could specifically recognize proteins in a phosphorylation-dependent manner, thereby allowing control of protein stability through protein kinase-mediated signal transduction.
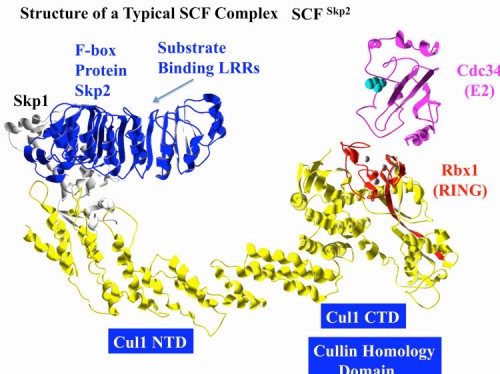
The SCF as the founding member of a large set of modular Cullin-based ligases
The initial discovery of Skp1 and F-box proteins identified related proteins known to exist in a second complex called the elongin complex that was originally thought to play a role in transcriptional elongation in vitro (77). Skp1 was closely related to elongin C, and the F-box was found to have sequence relatedness to the elongin A and Vhl1 tumor suppressor which both bind elongin C. This implicated this parallel complex in ubiquitination. Based on these findings, William Kaelin’s lab showed that the elongin complex also contains a Cullin (Cul2). This new modular ligase also acted as a ubiquitin ligase as demonstrated by the Conway lab. It is now known that each Cullin is a scaffold for a distinct modular ubiquitin ligase complex. Several of these complexes, such as the Cul3 ligase (168), lack a Skp1 subunit but instead fuse a Skp1-like domain, such as the BTB domain for Cul3, to a protein-protein interaction domain, essentially creating a Skp1-F-box protein fusion in order to recruit substrates. There are currently 7 cullin-based ligases, Cul1, Cul2, Cul3, Cul4A, Cul4B, Cul5 and Cul7. In addition, the anaphase promoting complex is a specialized Cullin ligase with three known specificity factors that contain WD40 repeats.
The RING Domain as an E2 recruitment domain revealed a large family of E3 ligases
Purification of Cul2 complexes by the Conaway lab identified a conserved RING domain protein we named Rbx1 (also known as Roc1), that our and Wade Harper’s labs showed to be required for SCF function in vitro and in vivo in yeast (110, 111). The RING domain on Rbx1 simultaneously bound Cul1 and an E2 and activated the E2 for ubiquitin transfer. Several other proteins implicated in ubiquitination also contained a RING domain and we hypothesized these proteins act as E3 recruit E2 enzymes to substrates. This has turned out to be the case. There are over 200 RING domain proteins in the human genome making it one of the largest classes of ubiquitin ligases.
F-box protein gene families in eukaryotes
Humans have aproximately 80 F-box proteins and an unknown number of equivalent adaptors for the other cullin ligases including BTB-domain proteins for Cul3, DCAF proteins for Cul4 and SOCS box proteins for CUL2 and CUL5. Since there is less known about the other adaptors, a conservative estimate is that these together with F-box proteins will comprise over 200 modular E3 ligases. In addition, each one of these has the ability to target multiple proteins for ubiquitination. BTRCP alone has nearly 40 known substrates. Thus, these ligases have the potential to regulate the stabilities of hundreds to thousands of proteins.
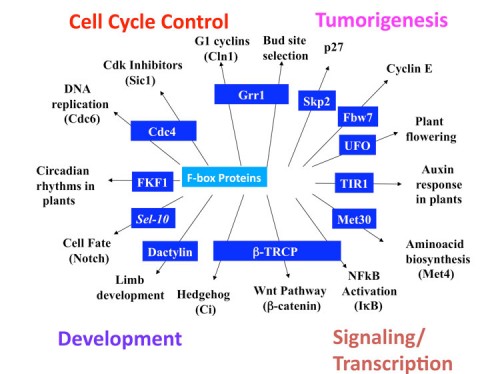
While humans have less than 100 F-box proteins, C. elegans is reported to have 300 and Arabidopsis has 700 F-box proteins. Furthermore, comparative genomics indicates that F-box proteins are the most rapidly evolving genes in Arabidopsis. This is likely due to the need of organisms like plants and worms to rapidly respond to their environment. While transcriptional responses ultimately remodel the transcriptome, controlling protein stability can occur on a much more rapid timescale, allowing a faster response to changing environmental stimuli which can mean the difference between life and death.
Identification of substrates for E3 ubiquitin ligases
A large number of substrates for the SCF have been identified, over 100 in humans, 40 in S. cerevisiae, and 21 in D. melanogaster have been matched with their cognate F-box. A current list of substrates are listed in these links for human (Link to SNAPSHOT II) and other species (Link to SNAPSHOT I). As can be seen, the substrates of the SCF alone comprise a large number of critical regulatory molecules including key cell cycle regulators, Cdk inhibitors, cyclins, DNA damage response proteins, cell polarity regulators, apoptosis regulators, growth factors, circadian regulators, inflammation regulators, immunoglobulin recombination etc. In most cases the substrate proteins are known to be key regulatory proteins and are linked to critical signal transduction pathways. The discovery of most of these substrates have come from analysis of the stability of a given protein, or by biochemical purification of F-box proteins looking for associated proteins under conditions when the destruction of the proteins are blocked. It is clear that identification of substrates of the Cullin ligases will reveal a biologically rich collection of proteins that underlie the physiological state of a cell.
Development of the GPS (global protein stability) system for the genome-wide measurement of protein stability
The abundance of cellular proteins is determined in large part by the rate of transcription and translation coupled with the stability of individual proteins. While we know a great deal about global transcript abundance, little is known about global protein stability. This deficiency has arisen because development of tools for a proteome-wide study of protein turnover is technologically challenging. There are more than six hundred E3s in the human genome, but we have functional information for only a small fraction of these. Many E3s have been shown to directly participate in human disease formation, and E3s are potentially effective targets of anticancer drugs. Regulated degradation of cancer-related proteins play important roles in cellular transformation, and multiple components of the proteolysis system are directly involved in human diseases. Therefore, development of proteome-wide approaches to deduce global protein stability profiling and E3-substrate networks are not only critical in furthering our understanding of normal protein turnover control and their deregulation in diseases, but also provide valuable information for the development of new therapeutic intervention strategies.
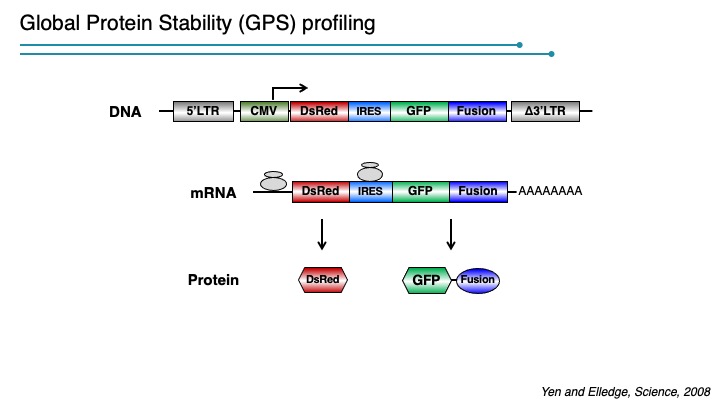
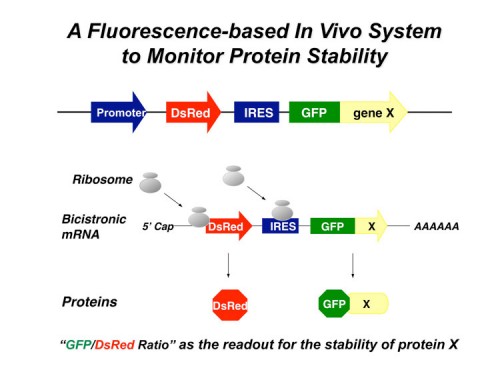
Traditional methods for measuring protein stability rely on either pulse-chase metabolic labeling or cycloheximide-chase analysis. However, these assays are impractical for the study of a large population of proteins under a broad range of physiological- or disease-states. An additional drawback of these methods is that they cannot be used to monitor protein turnover in living cells at single cell resolution, a feature important for a systems level understanding of protein function. To overcome these challenges, we established a live cell-based system for measuring global protein stability, GPS (217).
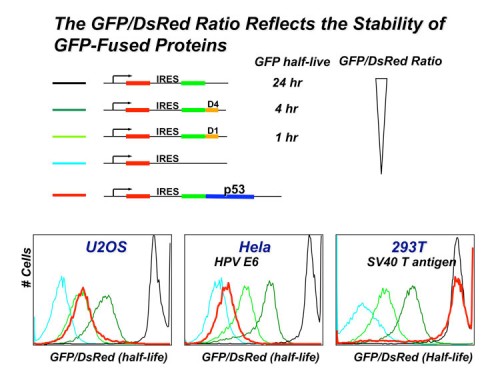
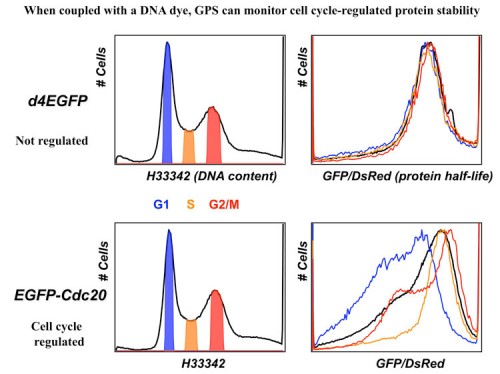
In this system, the expression cassette contains a single promoter that, with an internal ribosome entry site (IRES), permits the translation of two fluorescent proteins from one mRNA transcript. The first fluorescent protein (DsRed) is expressed as an intact protein and serves as an internal control, while the second fluorescent protein (EGFP) is expressed as a fusion protein with the protein of interest (EGFP-X). Upon integration into the genome of cells, DsRed and EGFP-X should be produced at a constant ratio since they are derived from the same mRNA, although their protein stabilities may differ. Events that selectively affect the protein stability of EGFP-X would be expected to change the abundance of EGFP-X, which should be reflected by an alteration of the EGFP/DsRed ratio. The EGFP/DsRed ratio serves as the stability readout in GPS and is not affected by transcription. When coupled with a dye such as Hoechst to measure DNA content, GPS can detect cel cycle regulated stability.
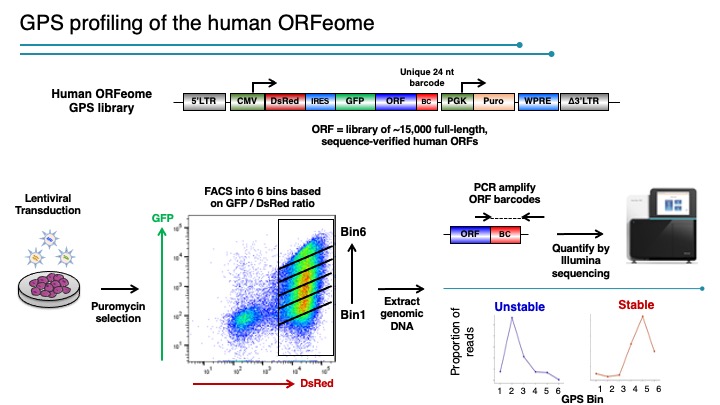
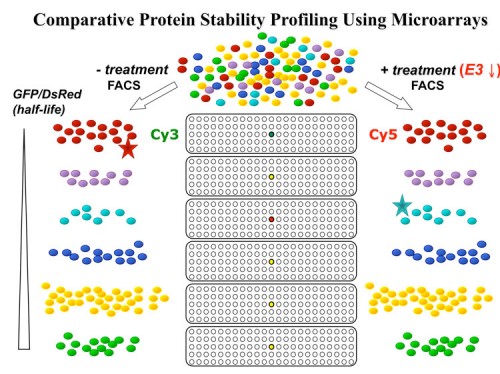
We designed a multiplexing strategy that coupled GPS with cDNA libraries and microarray deconvolution to profile the stability of ~15,000 human proteins in our latest experiment (218). This work represents the first example of large-scale protein stability measurements in human cells. Analysis of the Gene Ontology categories of stable and unstable proteins reveals a breakdown of classes of stabilities. Proteins with short half-lifes were enriched for cell communication, signal transduction, transport, oxidative phosphorylation and neuronal activities while long lived proteins are enriched for cell cycle, transcription, RNA splicing, DNA repair, DNA metabolism and exocytosis – activities cells are using and reusing every cell cycle. Based on our results, we think this technology can provide a general platform for proteome-scale analysis of protein turnover under various physiological and disease conditions.
Identification of Cul1 substrates
Linking an E3 with its substrates has been difficult and is generally dependent on either a functional connection or a physical association between the proteins. Performing biochemical screens has proven not an effective way to identify E3 substrates, as the binding between E3s and substrates is intrinsically weak. We applied GPS coupled with genetic ablation of E3 function to screen for the substrates of the SCF ubiquitin ligase in mammalian cells. We recovered most known SCF targets and generated a list of a few hundred proteins that are potential SCF substrates. Our results demonstrate that the GPS approach can provide a more effective and general solution for E3 substrate identification. Besides its use in studying ubiquitin-mediated proteolysis, this strategy can be further generalized to detect proteins whose stabilities increase or decrease in response to various stimuli such as, for example, cytokine stimulation, irradiation, and heat shock.
We have continued our exploration of the Cullin modular E3 ligase families and have now screened for substrates of Cul2, Cul3, Cul4 and Cul5 in addition to Cul1. We find that the sets of candidate protein substrates we identify for the different Cul proteins do not overlap. Importantly, most substrates of the SCF are critical regulators of their pathways thus, we anticipate that these substrates will also control many critical pathways and serve as a sensitive indicator of the physiological state of the cell to help provide a systems level understanding of the proteome for elucidation of regulation and disease mechanisms.


