Immunology Technology Development
Our lab is interested in developing new genetic technologies with applications for gene discovery and human health. Some of the technologies we are currently exploring are genetic screening with RNAI and ORF libraries, autoantibody discovery, and new methods for antibody discovery. Currently, we are particularly interested in auto-immune diseases and vaccine design. Some of our work is detailed below.
- Recent Immunological Methods
- T-Scan: A genome-wide method for the systematic discovery of T cell epitopes
- EpiScan: A synthetic biology platform for targeted immunopeptidomics
- VirScan: Comprehensive serological profiling of human populations using a synthetic human virome
- PICASSO: A CRISPR-based peptide library display and programmable microarray self-assembly for rapid quantitative protein binding assays
- V-CARMA: A method for viral infection of antigen-specific subsets of T cells
- AllerScan: High-resolution epitope conservation among peanut allergy patients detected by AllerScan reveals relationships between IgF and IgG repertoires during peanut oral immunotherapy
- Older Work
Recent Immunological Methods
T-Scan: A genome-wide method for the systematic discovery of T cell epitopes
T cell recognition of specific antigens mediates protection from pathogens and controls neoplasias, but can also cause autoimmunity. Our knowledge of T cell antigens and their implications for human health is limited by the technical limitations of T cell profiling technologies. To address this problem we developed T-Scan, a high-throughput platform for identification of antigens productively recognized by T cells.
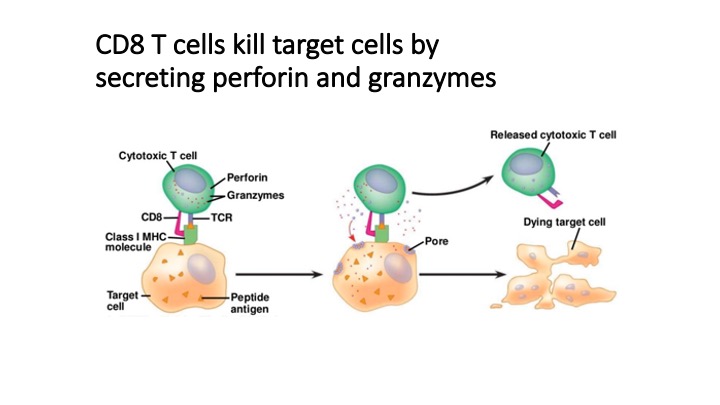
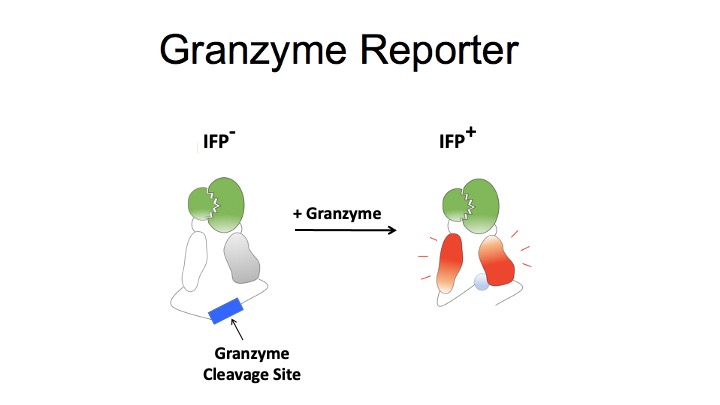
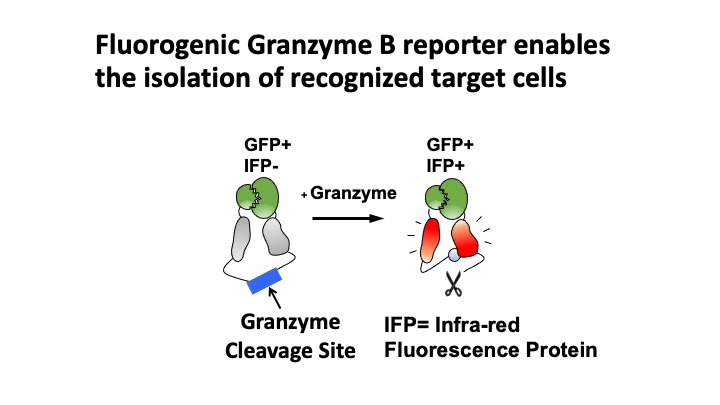
T-Scan uses lentiviral delivery of antigen libraries into cells for endogenous processing and presentation on MHC molecules. Target cells functionally recognized by T cells are isolated using a reporter for granzyme B activity and the antigens mediating recognition are identified by nextgen sequencing. T-Scan correctly identifies cognate antigens of T cell receptors (TCRs) from viral and human genome-wide libraries.
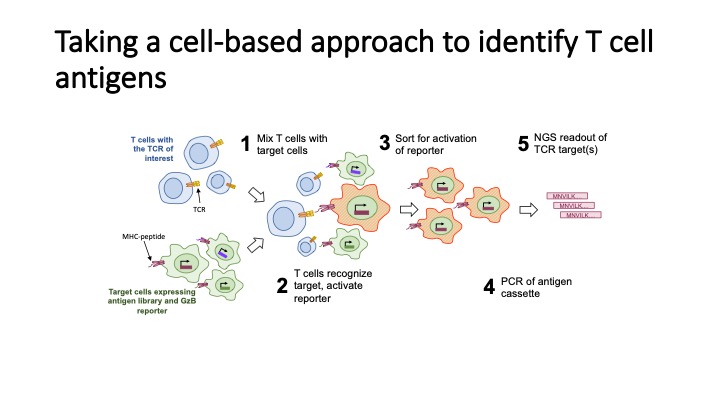
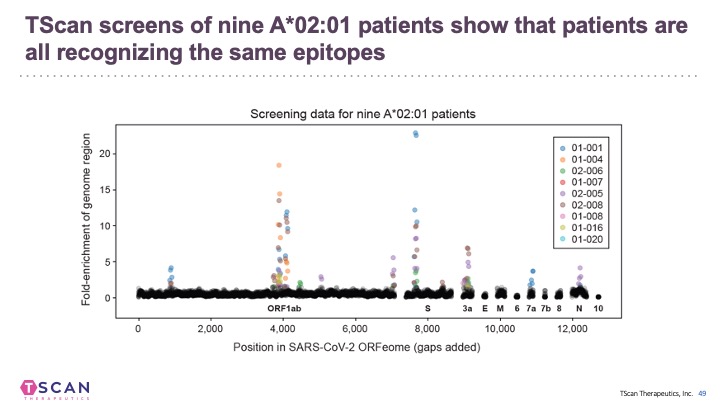
We and others have applied T-Scan to discover new viral antigens, perform high-resolution mapping of TCR specificity, and characterize the reactivity of a tumor-derived TCR. T-Scan is a powerful approach for studying T cell responses. T-Scan has also been used to identify immuno-dominant MHC-specific SARS-CoV2-derived peptide antigens from COVID-19 patients.
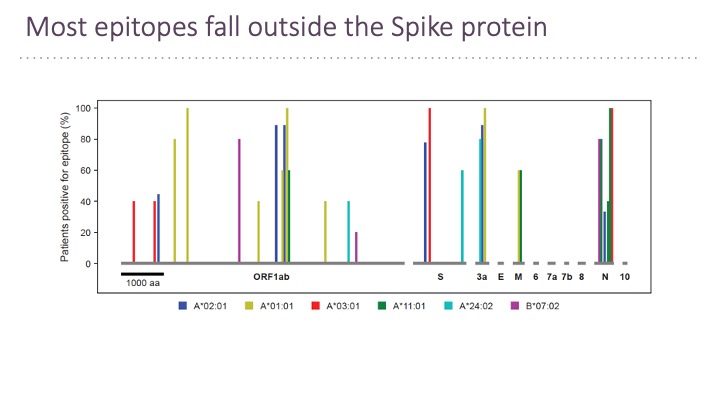
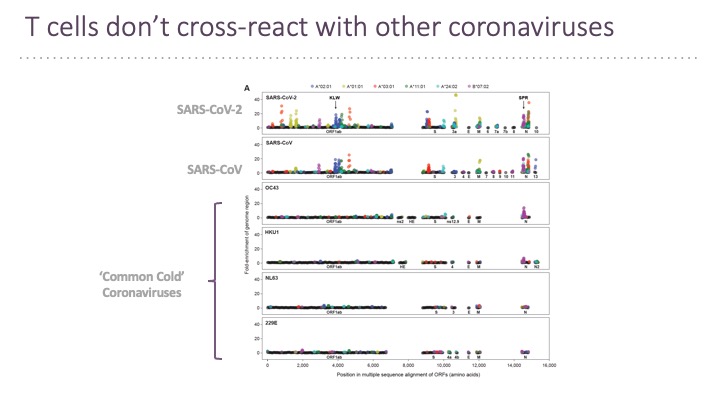
In addition to B cell epitopes, the TSCAN platform has been applied to patient T cells for T cell epitopes determination (Feretti et al, 2020 Immunity 53, 1095-1107.) T cell epitopes cover the entire virus at a frequency that corresponds to the ORF length and therefore few epitopes are in the Spike protein. Very few epitopes from SARS-CoV-2 cross-react with other human coronaviruses.
EpiScan: A synthetic biology platform for targeted immunopeptidomics
Identification of CD8+ T-cell epitopes is critical for the development of immunotherapeutics. Existing methods for MHC-I ligand discovery are time-intensive, specialized and unable to interrogate specific proteins on a large scale. To solve this problem we developed EpiScan, a programmable, high-throughput genetic method to identify MHC-I ligands amongst predetermined starting pools comprising >100,000 peptides.
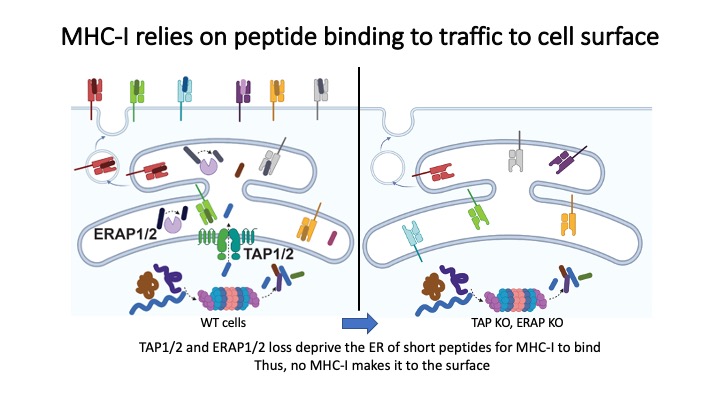
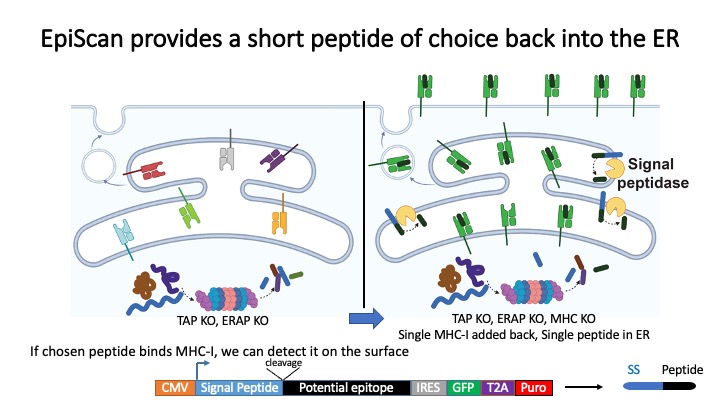
We exploit the programmability of EpiScan to uncover an unappreciated role for cysteine that increases the number of predicted ligands by ~22%, reveal affinity hierarchies by analysis of biased-anchor peptide libraries, and screen viral proteomes for MHC-I ligands. Using these data, we generate and iteratively refine peptide binding prediction program called ESP (EpiScan Predictor).
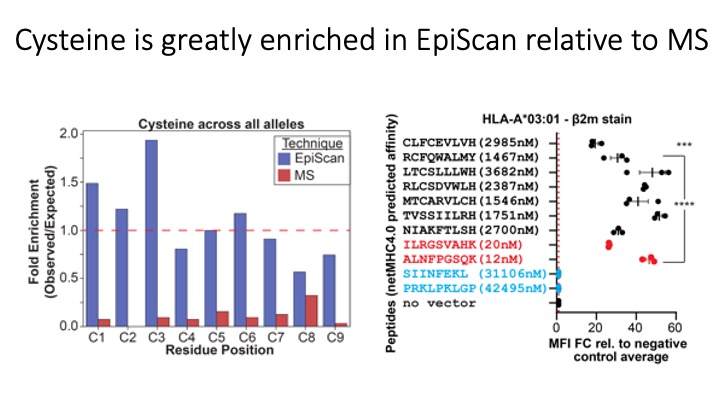
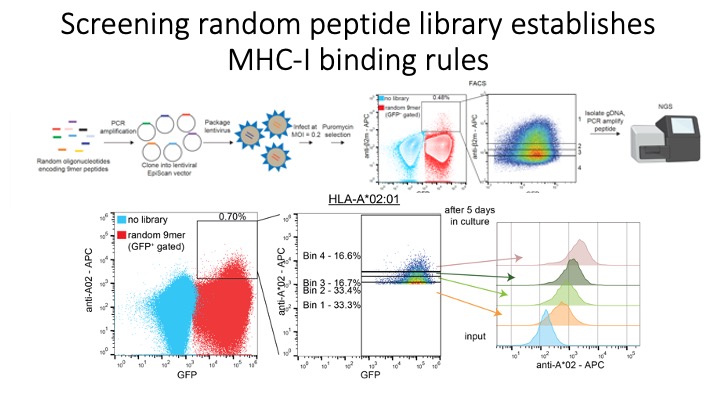
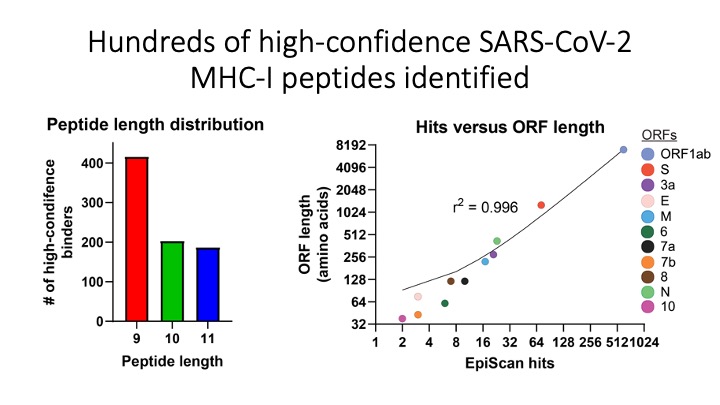
We programmed EpiScan to identify SARS-CoV-2 peptides that bind 11 different frequently inherited HLA alleles and showed that many of these peptides are antigens recognized by T cells. Targeted immunopeptidomics using EpiScan will accelerate CD8+ T-cell epitope discovery towards the goal of patient-specific immunotherapeutics.
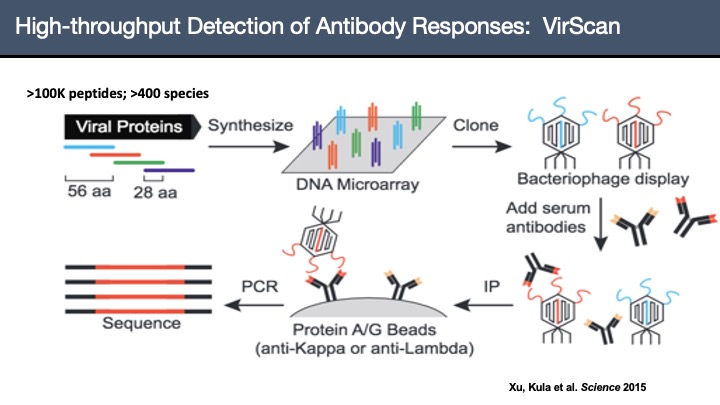
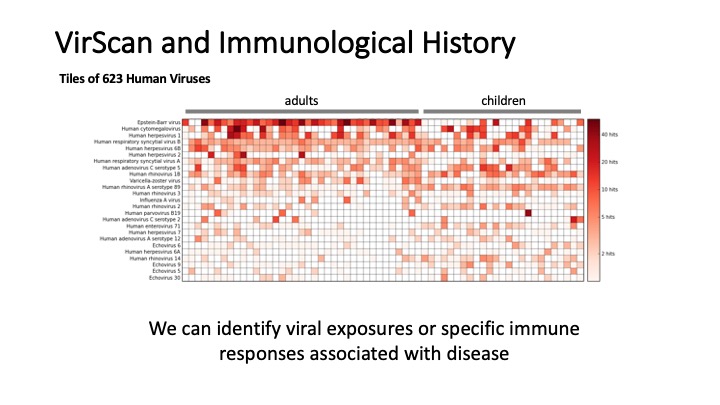
VirScan: Comprehensive serological profiling of human populations using a synthetic human virome
The human virome plays important roles in health and immunity. However, current methods for detecting viral infections and antiviral responses have limited throughput and coverage. To address this need we developed VirScan, a high-throughput method to comprehensively analyze antiviral antibodies using immunoprecipitation and massively parallel DNA sequencing of a bacteriophage library displaying proteome-wide peptides from all human viruses. We assayed over 108 antibody-peptide interactions in 569 humans across four continents, nearly doubling the number of previously established viral epitopes. We detected antibodies to an average of 10 viral species per person and 84 species in at least two individuals. Although rates of specific virus exposure were heterogeneous across populations, antibody responses targeted strikingly conserved “public epitopes” for each virus, suggesting that they may elicit highly similar antibodies. VirScan is a powerful approach for studying interactions between the virome and the immune system.

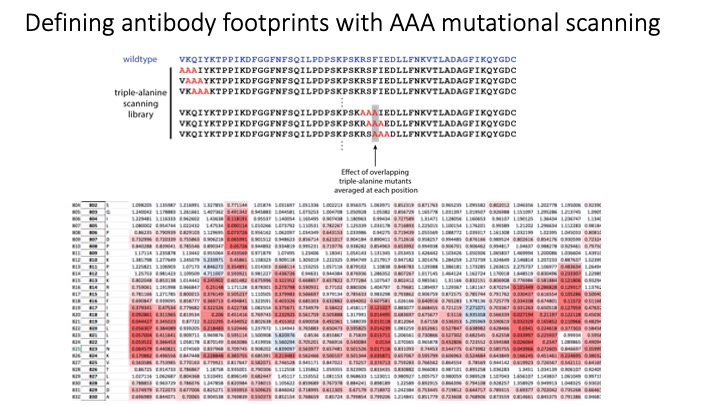
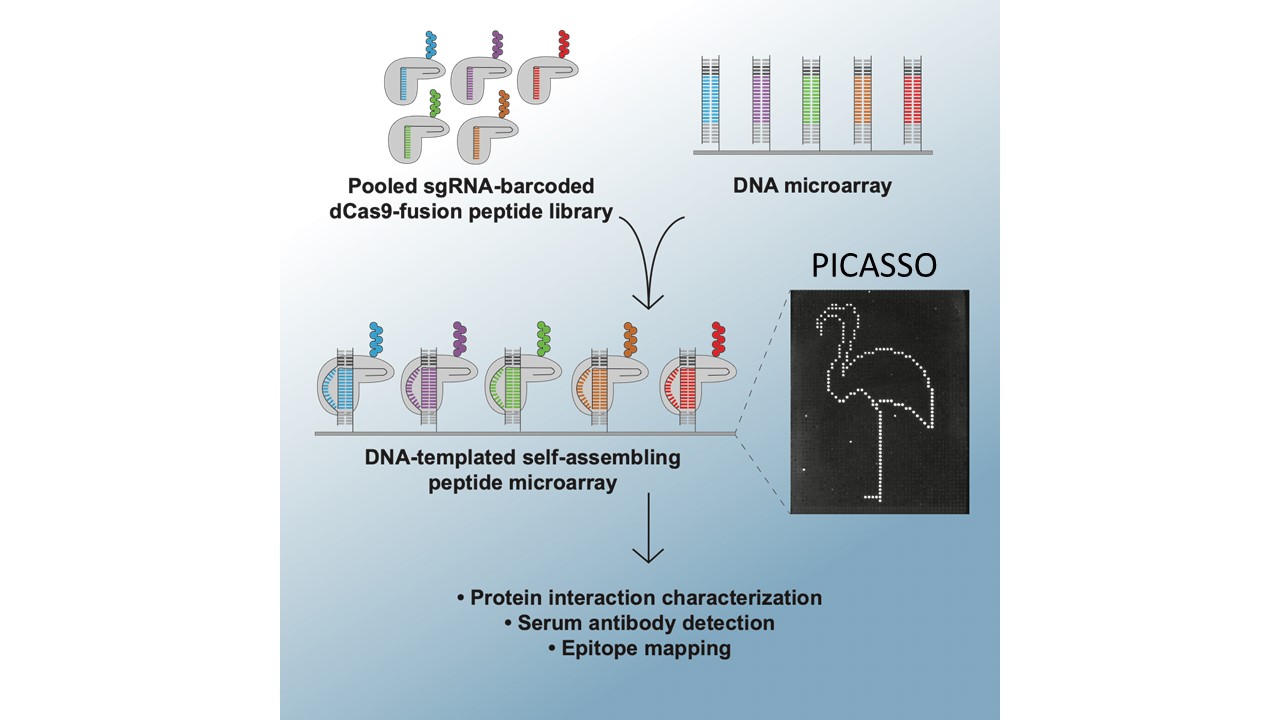
PICASSO: A CRISPR-based peptide library display and programmable microarray self-assembly for rapid quantitative protein binding assays
Microarray printing for nucleic acids has been a major advance for a number of applications, but the protein equivalent of these arrays has lagged behind. Like microarrays, CRISPR-inspired systems have been extensively developed for applications in genome editing and nucleic acid detection. To facilitate the study of custom peptide libraries and overcome the limitations of existing display technologies, we leveraged the properties of CRISPR systems to create a new in vitro peptide display platform. By fusing recombinant peptides to dCas9, we were able to barcode peptide libraries with unique identifier sgRNA sequences. Then, using a technique we call peptide immobilization by Cas9-mediated self-organization (PICASSO), the single mixed pool of dCas9-fusion peptides is able to localize to user-programmed positions on a microarray surface containing DNA sequences complementary to each peptide’s sgRNA barcode. We demonstrated that bespoke peptide libraries fused to catalytically inactive Cas9 (dCas9) and barcoded with unique single guide RNA (sgRNA) molecules self-assemble from a single mixed pool to programmable positions on a DNA microarray surface for rapid, multiplexed binding assays. We develop dCas9-displayed saturation mutagenesis libraries to characterize antibody-epitope binding for a commercial anti-FLAG monoclonal antibody and human serum antibodies. We also showed that our platform can be used for viral epitope mapping and exhibits promise as a multiplexed diagnostics tool. This is the first demonstration of CRISPR-based peptide display as well as complex library self-assembly using dCas9. This platform could be adapted for rapid interrogation of varied customized protein libraries or biological materials assembly using DNA scaffolding.
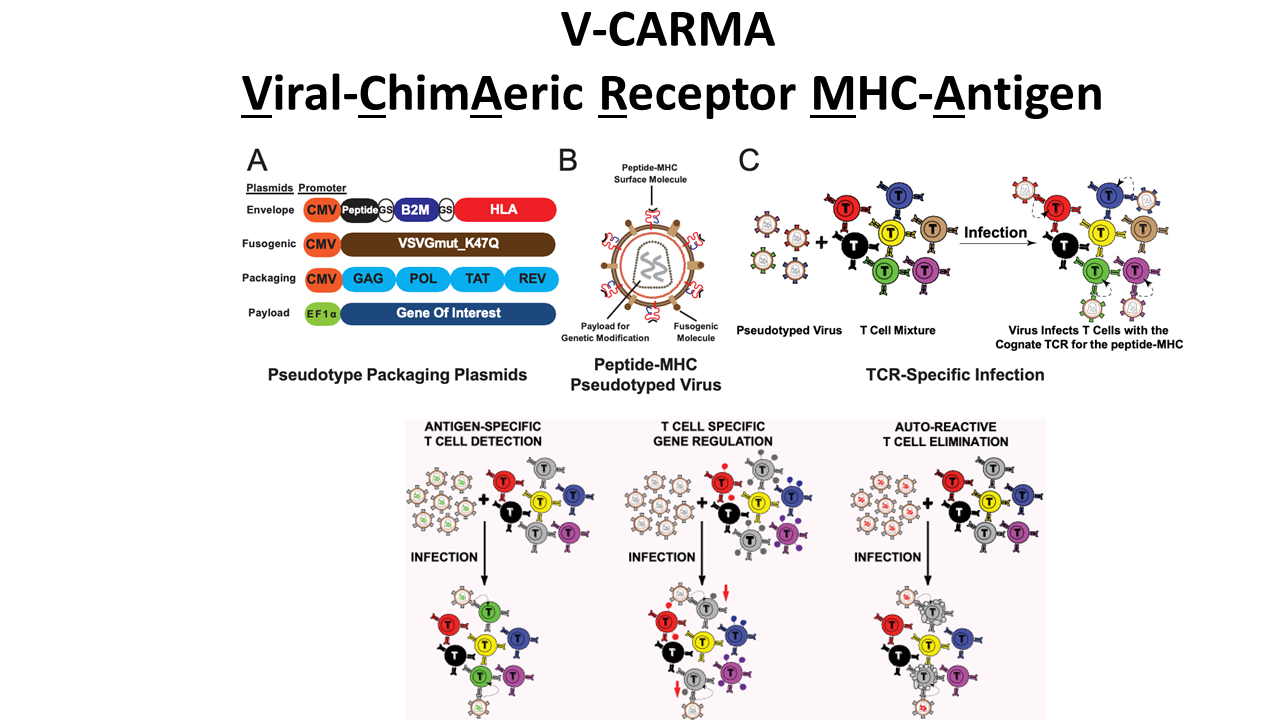
V-CARMA: A method for viral infection of antigen-specific subsets of T cells
T cells promote our body’s ability to battle cancers and infectious diseases but can act pathologically in autoimmunity. The recognition of antigens presented by major histocompatibility complexes (pMHC) molecules by T cell receptors (TCRs) enables T cell-mediated responses. To modify disease-relevant T cells, new tools to genetically modify T cells and decode their antigen recognition are needed.
To address this need we developed V-CARMA (Viral ChimAeric Receptor MHC-Antigen) an approach using viruses pseudotyped with peptides loaded on MHC to specifically target T cells expressing cognate TCRs for antigen discovery and T cell engineering. We found that lentiviruses displaying antigens on human leukocyte antigen (HLA) class I and class II molecules can robustly infect CD8+ and CD4+ T cells expressing cognate TCRs, respectively. The infection rates of the pseudotyped lentiviruses are directly correlated with the binding affinity of the TCR to its cognate antigen. Furthermore, peptide-HLA pseudotyped lentivirus V-CARMA constructs can identify target cells from a mixed T cell population, suppress PD-1 expression on CD8+ T cells via PDCD1 shRNA delivery, and induce apoptosis in auto-reactive CD4+ T cells. Thus, V-CARMA is a versatile tool for TCR ligand identification and selective T cell manipulation.
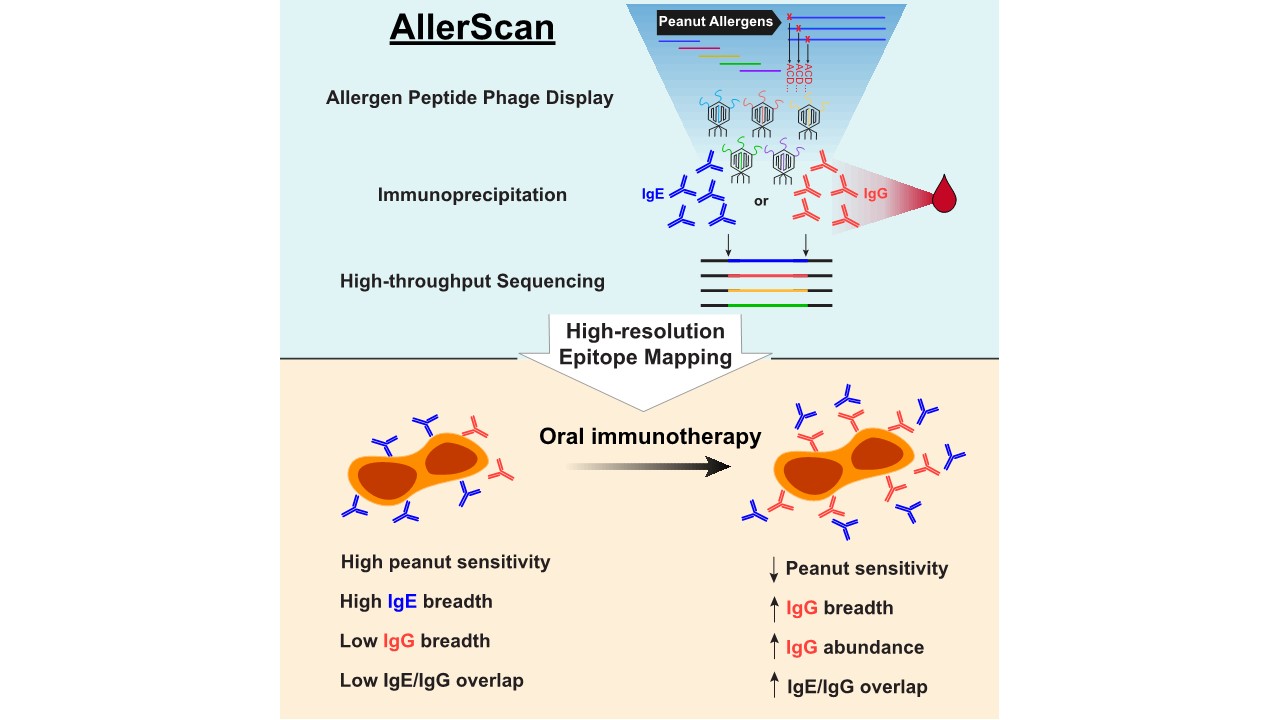
AllerScan: High-resolution epitope conservation among peanut allergy patients detected by AllerScan reveals relationships between IgE and IgG reertoires during peanut oral immunotherapy
Peanut allergy can result in life-threatening reactions and is a major public health concern. Oral immunotherapy (OIT) induces desensitization to food allergens through administration of increasing amounts of allergen.

To dissect peanut-specific immunoglobulin E (IgE) and IgG responses in subjects undergoing OIT, we have developed AllerScan, a method that leverages phage-display and next-generation sequencing to identify the epitope targets of peanut-specific antibodies. We observe a striking diversification and boosting of the peanut-specific IgG repertoire after OIT and a reduction in pre-existing IgE levels against individual epitopes. High-resolution epitope mapping reveals shared recognition of public epitopes in Ara h 1, 2, 3, and 7. In individual subjects, OIT-induced IgG specificities overlap extensively with IgE and exhibit strikingly similar antibody footprints, suggesting related clonal lineages or convergent evolution of peanut-specific IgE and IgG B cells. Individual differences in epitope recognition identified via AllerScan could inform safer and more effective personalized immunotherapy.
Older Work
shRNA Libraries
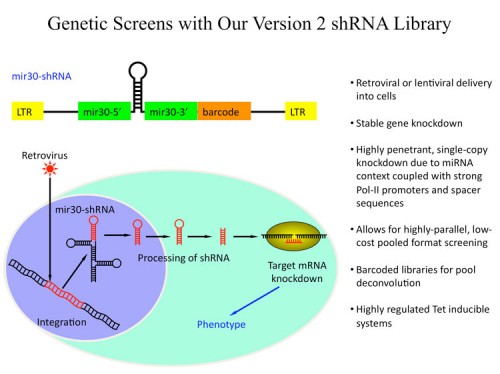
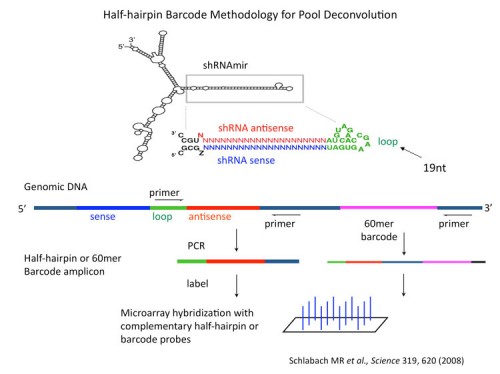
We, together with our collaborators in Greg Hannon’s laboratory, have generated large libraries of shRNAs that cover the entire human and mouse genomes (171, 183). The libraries are designed using shRNA prediction algorithms and synthesized in a massively parallel fashion using Agilent microarrays. Oligonucleotides are PCR amplified and cloned into retroviral or lentiviral vectors. At this point they can either be used directly or sequence verified and arrayed. Each vector also has a unique barcode associated with it for microarray deconvolution of pools. In this way we can generate large libraries of shRNAs for genetic screening (see the Technology Development Section for more details).
New shRNA vectors, the PRIME Vectors
The advent of RNAi has led to the ability to interfere with gene expression and greatly expanded our ability to perform genetic screens in mammalian cells. The expression of short hairpin RNA from polymerase III promoters can be encoded in transgenes and used to produce siRNAs that downregulate specific genes. However, we discovered that polymerase II-transcribed shRNAs display very efficient knockdown of gene expression when the shRNA is embedded in a micro-RNA context (182). Importantly, our shRNA expression system (called PRIME vectors) allows for the multicistronic co-transcription of a reporter gene, thereby facilitating the tracking of shRNA production in individual cells. Based on this system, we developed a series of lentiviral vectors that display tetracycline-responsive knockdown of gene expression at single copy. The high penetrance of these PRIME vectors will facilitate genome-wide loss-of function screens and is an important step towards using bar coding strategies to follow loss of specific sequences in complex populations.

We have also developed a series of PRIME vectors that show better knockdown in mouse ES cells and other cells where the CMV promoter might not be optimal. In addition, we have developed more tet-responsive inducible vectors that encode their own reverse tet-activator.
Recombinant DNA Technologies
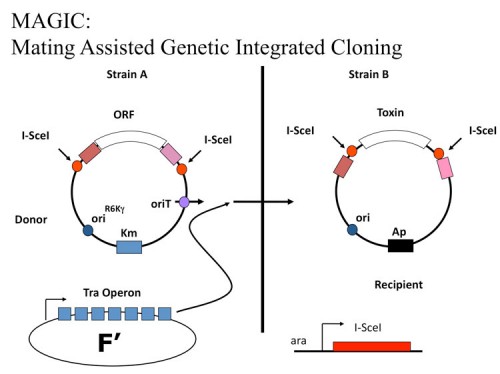
MAGIC: An in vivo genetic method for the rapid construction of recombinant DNA
We developed a novel, highly engineered in vivo cloning method, MAGIC (Mating Assisted Genetically Integrated Cloning), that facilitates the rapid construction of recombinant DNA molecules (180). MAGIC employs bacterial mating, in vivo site-specific endonuclease cleavage and homologous recombination to catalyze DNA fragment transfer between a donor vector in one bacterial strain and a recipient plasmid in a separate bacterial strain. Recombination events are genetically selected and result in placement of the gene of interest under the control of novel regulatory elements with high efficiency. MAGIC eliminates the need for restriction enzymes, DNA ligases, preparation of DNA and all in vitro manipulations required for subcloning and allows the rapid construction of multiple constructs with minimal effort. We demonstrate MAGIC can generate constructs for expression in multiple organisms. As this new method requires only the simple mixing of bacterial strains, it represents a significant advance in high throughput recombinant DNA production that will save researchers significant amounts of time, effort and expense in functional genomics studies.
SLIC Cloning: a method for subcloning without restriction enzymes or DNA ligase
We developed a novel cloning method SLIC (Sequence and Ligation-Independent Cloning) that allows the assembly of multiple DNA fragments in a single reaction using in vitro homologous recombination and single-strand annealing (195). SLIC mimics in vivo homologous recombination by relying on exonuclease generation of single strand DNA (ssDNA) overhangs on insert and vector fragments and the assembly of these fragments by recombination in vitro. SLIC inserts can be prepared by incomplete PCR (iPCR) or mixed PCR. SLIC allows efficient and reproducible assembly of recombinant DNA with as many as 5 and 10 fragments simultaneously. SLIC circumvents the sequence requirements of traditional methods and is much more sensitive when combined with RecA to catalyze homologous recombination. It also provides a new method for site-directed mutagenesis of a gene. This flexibility allows much greater versatility in the generation of recombinant DNA for the purposes of synthetic biology.

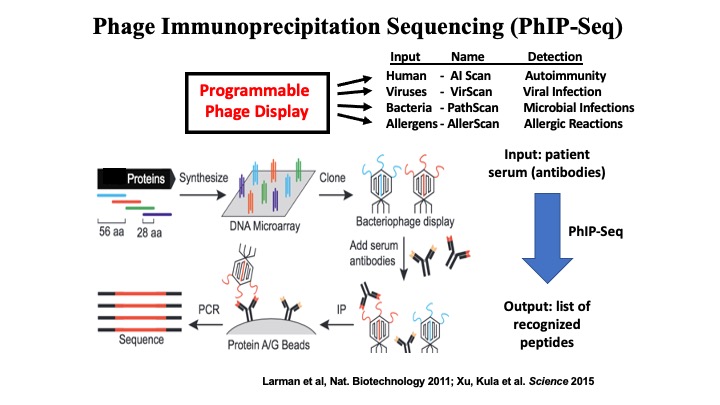
PhIP Seq: Enhanced display technologies for biomarker discovery
The sensitive detection of circulating biomarkers represents a powerful approach to screening for early malignancy. However, current techniques are limited in their ability to distinguish normal from pathological states. This work aims to improve on current screening technology, utilizing novel principles in synthetic biology. In particular, we combine next generation DNA sequencing with the display of polypeptide libraries encoded by microarray-derived oligonucleotides. Using the T7 bacteriophage system, we are able to display the complete human peptidome and thereby seek to discover autoantigen biomarkers common to breast cancer patients. In parallel we are developing a fully defined, synthetic human single chain variable fragment (scFv) antibody library. This library will be used in ribosome display format for a scFv library-versus-peptide library screen in an effort to create a proteomic scale scFv set. Our hope is that development of these technologies will facilitate the identification of circulating cancer biomarkers, thereby enabling early diagnosis and treatment of the disease.
The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo
The discovery of RNAi has revolutionized loss-of-function genetic studies in mammalian systems. However, significant challenges still remain to fully exploit RNAi for mammalian genetics. For instance, genetic screens and in vivo studies could be broadly improved by methods that allow inducible and uniform gene expression control. To achieve this, we built the lentiviral pINDUCER series of expression vehicles for inducible RNAi in vivo. Using a multicistronic design, pINDUCER vehicles enable tracking of viral transduction and shRNA or cDNA induction in a broad spectrum of mammalian cell types in vivo. They achieve this uniform temporal, dose-dependent, and reversible control of gene expression across heterogenous cell populations via fluorescence-based quantification of reverse tet-transactivator expression. This feature allows isolation of cell populations that exhibit a potent, inducible target knockdown in vitro and in vivo that can be used in human xenotransplantation models to examine cancer drug targets.
Lambda YES cDNA Expression System
We have developed a multifunctional lambda expression vector system, lambda YES, designed to facilitate gene isolation from eukaryotes by complementation of E. coli and S. cerevisiae mutations (23). Lambda YES vectors have a selection for cDNA inserts using an oligo adaptor strategy and are capable of expressing genes in both E. coli and S. cerevisiae. They also allow conversion from phage l to plasmid clones using the cre-lox site-specific recombination system, referred to as automatic subcloning. Lambda YES vectors utilize an adaptor selection for inserts and can generate libraries with 109 recombinants per mg of cDNA insert. cDNA libraries constructed in these vectors have been used to isolate genes from humans by complementation of yeast. Using this technology we isolated the human Cdk2 gene (24) by complementation of a yeast cdc28 Ts mutation and the Cyclin F gene (40) as a suppressor of cdc4 Ts mutations which led to the discovery of F-box proteins (77).
UPS – the Univector Plasmid Fusion System: A method for rapid construction of recombinant DNA molecules without restriction enzymes
Modern biological research is highly dependent upon recombinant DNA technology. The functional analysis of a single gene requires the introduction of that gene into multiple vectors for different purposes. Conventional cloning methods are time-consuming and individual cloning events must be performed independently. To approach solving this problem, we sought to develop a systematic and uniform method with which to manipulate large sets of genes.
We developed a series of novel cloning methods that facilitate the rapid and systematic construction of recombinant DNA molecules (102). The central method is named the Univector Plasmid-fusion System (UPS). UPS employs cre-lox site-specific recombination to catalyze plasmid fusion between the Univector, a plasmid containing the gene of interest, and host vectors containing regulatory information. Fusion events are genetically selected and result in placement of the gene under the control of novel regulatory elements. A second UPS-related method allows for the precise transfer of coding sequences only from the Univector into a host vector. UPS eliminates the need for restriction enzymes, DNA ligases, and many in vitro manipulations required for subcloning and allows the rapid construction of multiple constructs for expression in multiple organisms. We demonstrate that UPS can be used to transfer whole libraries into new vectors. New methods for directional cloning of PCR fragments and for the generation of epitope tags and other fusions at the 3′ end of genes using homologous recombination in E. coli are described that facilitate cloning and manipulation of genes in the Univector.
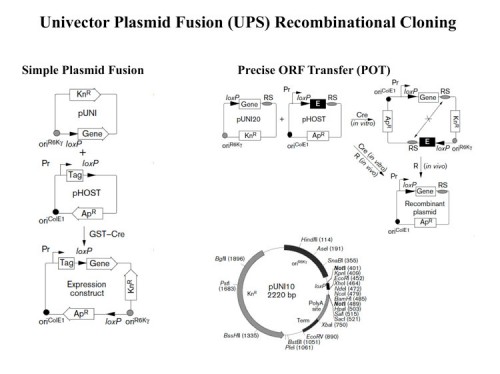
Together, these recombination-based cloning methods constitute a new comprehensive approach for the rapid and efficient generation of recombinant DNA that can be used for parallel processing of large gene sets, an ability required for future genomic analysis. We are continuing to develop large-scale capabilities using UPS as an alternative to the expensive and limited GATEWAY system.
Advances in the Two-Hybrid System
The two-hybrid cloning system is a powerful genetic method to identify genes via protein-protein interactions. We have made a number of improvements to this method which are designed to increase the number of potential protein-protein interactions detectable and to streamline the labor intensive process of authenticating clones identified in the primary screen. The first improvement was the design of genetic selections instead of screens to detect interacting proteins (32, 39). We developed the strain Y190 using HIS3 as a reporter and Y166 which employed a URA3 reporter for this purpose. A selection vastly increases the numbers of library clones that can be searched and is now built into all systems. Secondly, through the use of negative selections for plasmid loss coupled with a replica-mating strategy, we have developed a rapid method to distinguish between the genuinely interacting clones and the nonspecific background that is inherent to the system. Third, a lambda phage vector, lambda ACT2, was made that allows construction of highly complex, HA epitope-tagged, directional libraries of GAL4 activation domain-fused cDNAs that can be converted to plasmid form by in vivo cre-lox mediated site-specific recombination. We also introduced yeast mating of library clones to baits as a rapid way to both generate libraries and well as to counter screen against false positives. These methods have been incorporated into all current two-hybrid system kits. We used these methods to identify the human p21 and p57 Cdk inhibitors.
A Cytoplasmic Two Hybrid System, The SOS Recruitment System
In collaboration with Michael Karin’s lab, we have developed a novel two hybrid system for the cloning of genes involved in protein-protein interactions (84). This system, called the SOS Recruitment System, SRS, relies on the activation of the ras signalling pathway when a fusion protein to the ras guanine nucleotide exchange factor, SOS, is recruited to the inner surface of the plasma membrane by physical association with a second protein targeted to the same membrane by a myristylation sequence. When SOS is successfully recruited to the plasma membrane, it activates ras and bypasses the need for the endogenous exchange factor, Cdc25, an essential protein. This system allows protein-protein interactions to be detected in the cytoplasm. Furthermore, proteins that activate transcription can be used in this system giving it an advantage over the transcriptionally based two-hybrid system. We have developed a lambda-based expression vector, lambda MS-TRP for expression of myristylation-fused cDNA libraries an other useful vectors for this method. Visit our protocols page for more information


