Cell Cycle
Our lab has had a long history of studying the cell cycle in both yeast and mammals. We continue to study aspects of the mammalian cell cycle, both entry and exit. Currently we are interested in identifying genes that control cell proliferation as well as understanding how cells permanently exit the cell cycle through a process of cellular senescence. Some of our work is described below.
- Recent Studies on Senescence
- The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4
- Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program
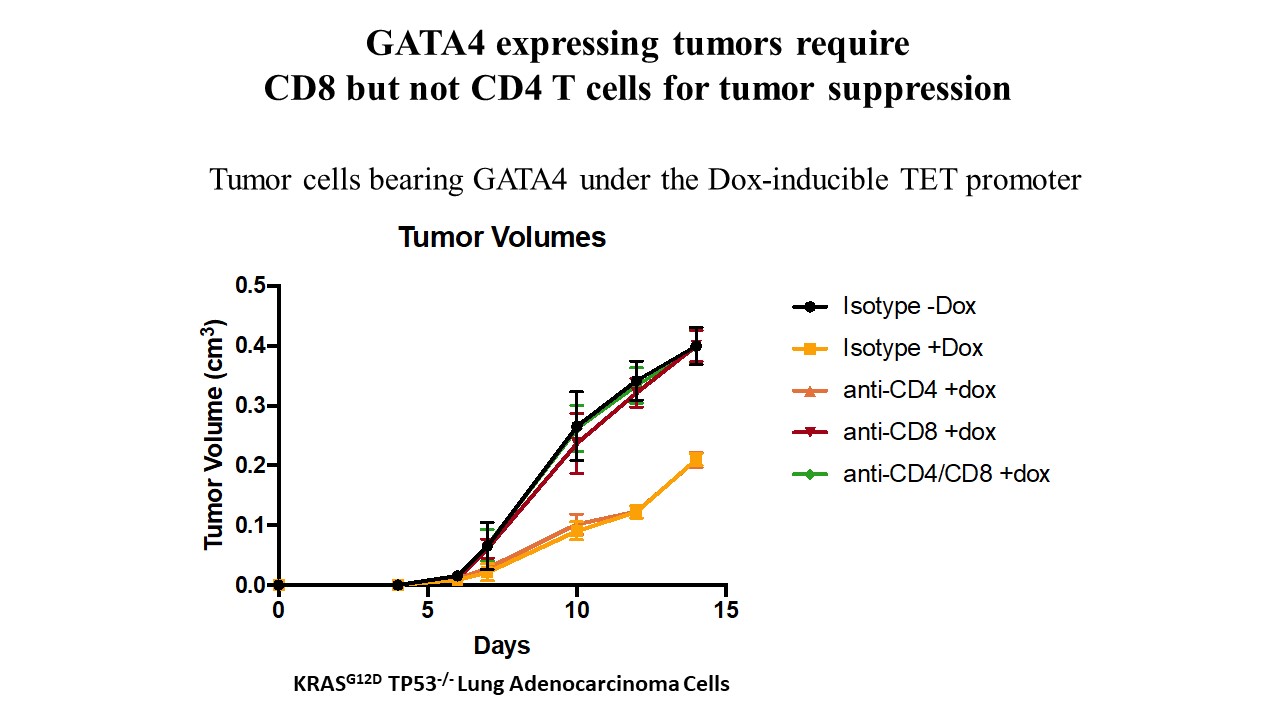
- A GATA4-regulated secretory program suppresses tumors through recruitment of cytotoxic CD8 T cells
- Older Work
Recent Studies
The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4
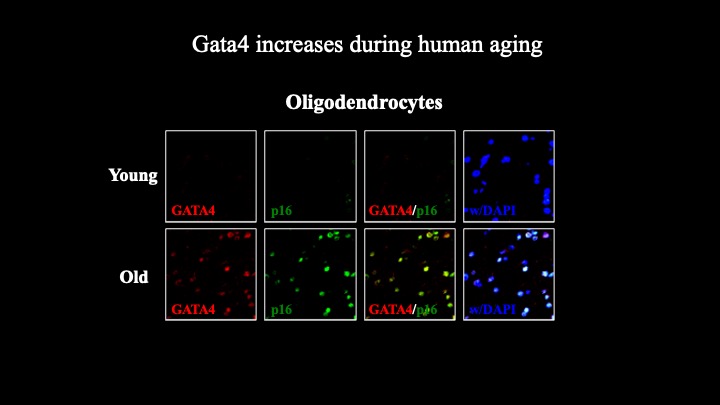
Cellular senescence is a terminal stress-activated program controlled by the p53 and p16(INK4a) tumor suppressor proteins. A striking feature of senescence is the senescence-associated secretory phenotype (SASP), a pro-inflammatory response linked to tumor promotion and aging. We have identified the transcription factor GATA4 as a senescence and SASP regulator. GATA4 is stabilized in cells undergoing senescence and is required for the SASP. Normally, GATA4 is degraded by p62-mediated selective autophagy, but this regulation is suppressed during senescence, thereby stabilizing GATA4. GATA4 in turn activates the transcription factor NF-κB to initiate the SASP and facilitate senescence. GATA4 activation depends on the DNA damage response regulators ATM and ATR, but not on p53 or p16(INK4a). GATA4 accumulates in multiple tissues, including the aging brain, and could contribute to aging and its associated inflammation.
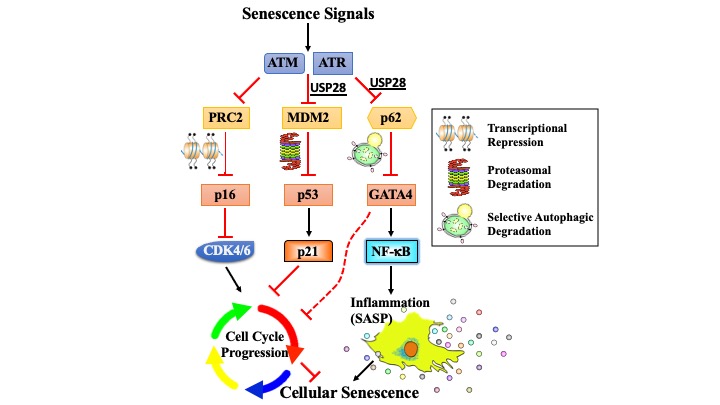
Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program
Senescence is a terminal differentiation program that halts the growth of damaged cells and must be circumvented for cancer to arise. Here we describe a panel of genetic screens to identify genes required for replicative senescence. We uncover a role in senescence for the potent tumor suppressor and ATM substrate USP28. USP28 controls activation of both the TP53 branch and the GATA4/NFkB branch that controls the senescence-associated secretory phenotype (SASP). These results suggest a role for ubiquitination in senescence and imply a common node downstream from ATM that links the TP53 and GATA4 branches of the senescence response.
A GATA4-regulated secretory program suppresses tumors through recruitment of cytotoxic CD8 T cells
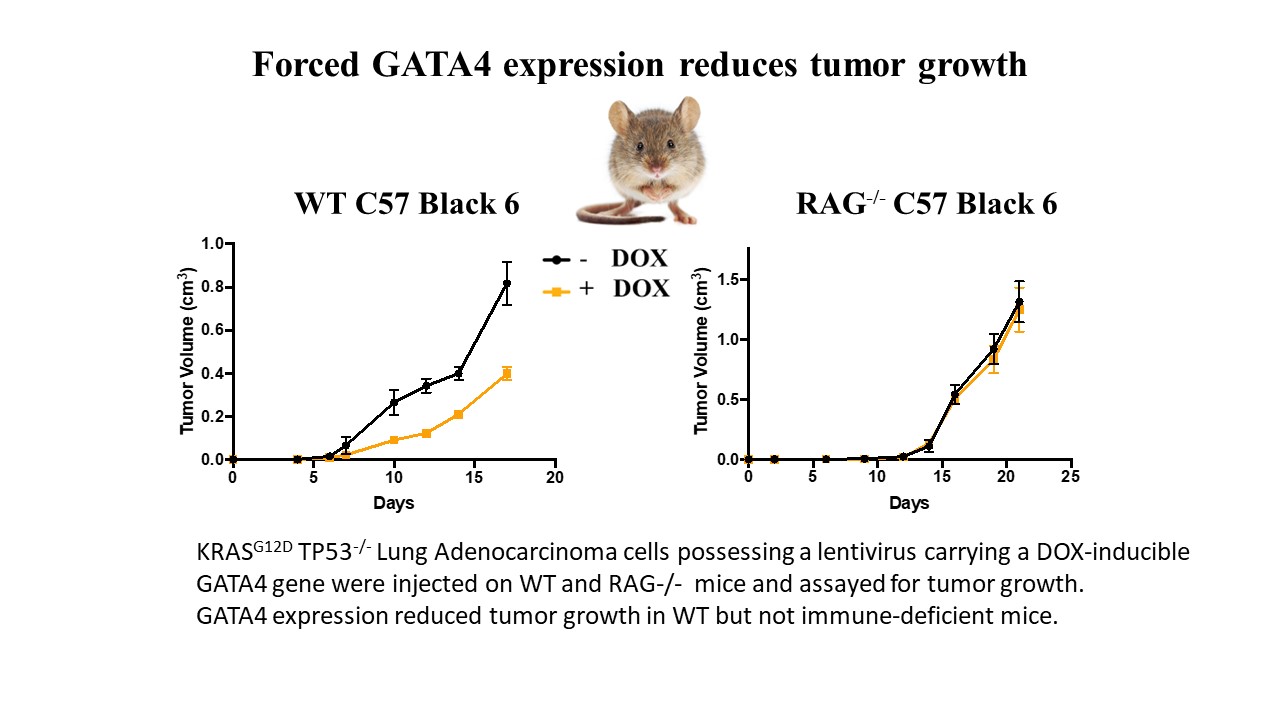
The GATA4 transcription factor acts as a master regulator of development of multiple tissues. GATA4 also acts in a distinct capacity to control a stress-inducible pro-inflammatory secretory program that is associated with senescence, a potent tumor suppression mechanism, but also operates in non-senescent contexts such as tumorigenesis. This secretory pathway is composed of chemokines, cytokines, growth factors, and proteases. Since GATA4 is deleted or epigenetically silenced in cancer, here we examine the role of GATA4 in tumorigenesis in mouse models through both loss-of-function and overexpression experiments. We find that GATA4 promotes non-cell autonomous tumor suppression in multiple model systems. Mechanistically, we show that GATA4-dependent tumor suppression requires cytotoxic CD8 T cells and partially requires the secreted chemokine Ccl2. Analysis of transcriptome data in human tumors reveals reduced lymphocyte infiltration in GATA4-deficient tumors, consistent with our murine data. Notably, activation of the GATA4-dependent secretory program combined with immune checkpoint inhibition with an anti-PD-1 antibody robustly abrogates tumor growth in vivo.
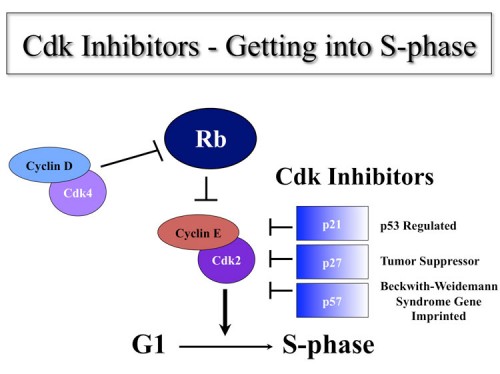
Older Work
Discovery of Cdk Inhibitors
A central theme of the research in this laboratory focuses on the identification and regulation of genes important for progression through the eukaryotic cell cycle. We are interested in how human cells decide when to synthesize DNA. Using novel expression strategies, we have identified a protein kinase, Cdk2, which appears to be the central cell cycle kinase that binds cyclin A and cyclin E and regulates entry into S-phase (24, 26, 30). We have pursued the regulation of Cdk2 and together with Dr. Wade Harper identified a novel class of cell cycle regulators of Cdks called CKIs, Cdk inhibitors. The first of which was p21CIP (39). There are now three members of the p21 family, p21, p27 and p57 (61) discovered in our lab and others. Work from our lab and others have determined that p21 is transcriptionally regulated by p53, is induced in response to DNA damage and controls the G1 checkpoint in response to DNA damage (59, 66). We mapped p57 to 11p15.5 which is the location of a number of imprinted genes and the site of a familial cancer syndrome BWS (Beckwith-Weidemann’s Syndrome). BWS patients often show uniparental disomy for the paternal region of 11p15.5. We found p57 is genomically imprinted (72). By insitu hybridization we found that p57 was highly expressed in the tissues involved in the phenotype of BWS patients. We found mutations in the human p57 gene in BWS patients (91) and made a knockout mouse that recapitulated the organ overgrowth phenotypes of these patients (85). Subsequence work we did in collaboration with Shirley Tighlman’s lab showed that loss of p57 coupled with overproduction of IGF2 which also localized to 11p15.5 but which has opposite imprinting provided an even better model for BWS disease (119).
We have explored the role of these proteins in the control of cell proliferation during mouse development and have found that they have both unique roles and redundant roles. p21 deficient mice are developmentally normal (66). p27 deficient mice display overgrowth and multi-organ hyperplasia. p57 has a role in the control of cell proliferation in limb growth, palate formation and in the lens (91). p21 and p57 play a redundant role in lung and muscle differentiation in vivo (105). p27 and p57 play redundant roles in lens development (103). Each of these have been implicated in tumorigenesis.
Cell cycle phosphoproteomics – a quantitative atlas of cell cycle regulated phosphorylation
The eukaryotic cell division cycle is characterized by a sequence of orderly and highly regulated events resulting in the duplication and separation of all cellular material into two newly formed daughter cells. Protein phosphorylation by cyclin-dependent kinases (CDKs) drives this cycle. To gain further insight into how phosphorylation regulates the cell cycle, we sought to identify proteins whose phosphorylation is cell cycle regulated (210). Using stable isotope labeling along with a two-step strategy for phos-phopeptide enrichment and high mass accuracy mass spectrometry, we examined protein phosphorylation in a human cell line arrested in the G1 and mitotic phases of the cell cycle. We identified >14,000 different phosphorylation events, more than half of which, to our knowledge, have not been described in the literature, along with relative quantitative data for the majority of these sites. We observed >1,000 proteins with increased phosphorylation in mitosis including many known cell cycle regulators. The majority of sites on regulated phosphopeptides lie in [S/T]P motifs, the minimum required sequence for CDKs, suggesting that many of the proteins may be CDK substrates. Analysis of non-proline site-containing phosphopeptides identified two unique motifs that suggest there are at least two undiscovered mitotic kinases.
Growth control in mammals
Using both gain of function and loss of function genetic screening, we are currently probing growth control in mammalian cells grown in culture. Our loss of function screens employ our shRNA technologies. Our gain of function screens now cover over 15,000 full length ORFs in viral vectors. We are applying these libraries to identify the underlying circuitry that governs how cells divide and how they circumvent stressors that are normally deleterious to their proliferation.
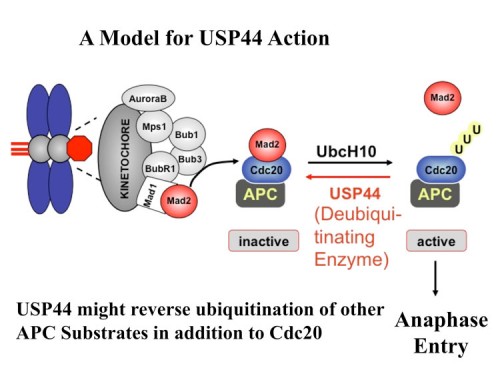
Anaphase initiation in mammals is regulated by antagonistic ubiquitination and deubiquitination activities
The spindle checkpoint prevents missegregation by delaying sister chromatid separation until all chromosomes have achieved bipolar attachment to the mitotic spindle. Its operation is essential for accurate chromosome segregation, while its dysregulation can contribute to birth defects and tumorigenesis. The target of the spindle checkpoint is the anaphase-promoting complex (APC), a ubiquitin ligase that promotes sister chromatid separation and progression to anaphase. Using an shRNA screen targeting components of the ubiquitin-proteasome pathway, we identified the deubiquitinating enzyme USP44 as a critical regulator of the spindle checkpoint (199). USP44 is not required for the initial recognition of unattached kinetochores and the subsequent recruitment of checkpoint components. Instead, it prevents the premature activation of the APC by stabilizing the APC-inhibitory Mad2-Cdc20 complex. USP44 deubiquitinates the APC-coactivator Cdc20 both in vitro and in vivo, and thereby directly counteracts the APC-driven disassembly of Mad2-Cdc20 complexes (see accompanying manuscript by Reddy et al.). Our findings suggest that a dynamic balance of ubiquitination by the APC and deubiquitination by USP44 contributes to the generation of the switch-like transition controlling anaphase entry analogous to the way that phosphorylation and dephosphorylation of Cdk1 by Wee1 and Cdc25 controls entry into mitosis.